Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
The inflammatory protease caspase-1 is associated with the release of cytokines. An excessive number of cytokines (a “cytokine storm”) is a dangerous consequence of COVID-19 infection and has been indicated as being among the causes of death by COVID-19. The anti-inflammatory drug colchicine (which is reported in the literature to be a caspase-1 inhibitor) and the corticosteroid drugs, dexamethasone and methylprednisolone, are among the most effective active compounds for COVID-19 treatment.
- COVID-19
- caspase-1
- cytokine storm
- anti-inflammatory drugs
1. Introduction
The mechanism of COVID-19 development remains unclear, although unregulated cytokine release resulting in an excessive inflammatory response is likely implicated. Several drugs previously used to modulate immune responses in many diseases, including two readily available and inexpensive corticosteroids, dexamethasone and methylprednisolone, have now been repurposed and used as promising new tools for reducing the inflammatory response to COVID-19 virus infection [1,2]. “Cytokine storm” and cytokine release syndrome are life-threatening systemic inflammatory syndromes involving elevated levels of circulating cytokines and immune-cell hyperactivation that can be triggered by various therapies, pathogens, cancers, autoimmune conditions and monogenic disorders [3]. These damaging conditions prompt the formation of inflammasomes, large multi-protein complexes that include the protease caspase-1. Caspase-1 cleaves interleukins, pro-IL-1β and pro-ΙL-18, into their operative cytokine forms, and also cleaves the protein gasdermin D, which triggers pyroptosis, a pro-inflammatory form of cell death. These are likely implicated in acute COVID-19 pathogenesis and may be more damaging to the host than the response associated with viral-induced cell death in the host [4]. An intensified immune response may be a major contributor to the poor outcomes often observed in patients with COVID-19 [5]. Figure 1, taken from [6], shows the mechanism of inflammasome activation.
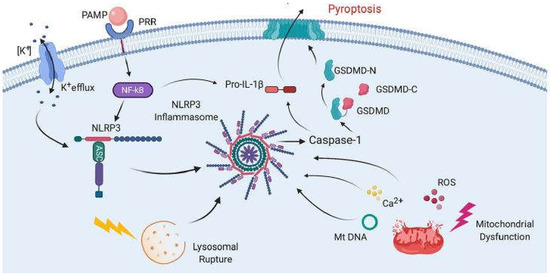
Figure 1. Mechanisms of inflammasome activation. The activation in general relies on two signals. First, pathogen-associated molecular patterns (PAMPs) bind to pattern recognition receptors (PRRs), resulting in the synthesis of NLRP3 and pro-IL-1β. Then, a second signal causes inflammasome assembly, leading to the activation of caspase-1 and the processing of pro-IL-1β into IL-1β and pyroptosis. The second signal may come from a variety of pathways, including K+ efflux, lysosomal rupture or mitochondrial dysfunction. This results in the release of ROS, Ca2+ and mitochondrial DNA (mtDNA), all of which have been shown to activate the inflammasome. Pyroptosis occurs as a result of caspase-1-mediated cleavage of GSDM-D at the linker region of GSDM-D. Following GSDM-D cleavage, the amino terminus of GSDM-D (GSDM-D-N) forms a non-selective pore at the cell membrane through which IL-1β is then released. (From [6], with permission.).
Activation of the inflammasome is a key function mediated by the innate immune system [7]. Several families of PRRs are important components in the inflammasome complex, including the nucleotide-binding domain, leucine-rich repeat containing proteins (NLRs, also known as NOD-like receptors). Upon sensing certain stimuli, the relevant NLR can oligomerize to be a caspase-1-activating scaffold. Active caspase-1 subsequently functions to cleave the proinflammatory IL-1 family of cytokines into their bioactive forms, IL-1β and IL-18, and cause pyroptosis, a type of inflammatory cell death [8,9].
The inhibition of caspase-1, an enzyme involved in triggering the release of pro-inflammatory cytokines, is relevant to the mechanism of anti-COVID-19 therapy [6]. Since there is a strong suspicion that excessive amounts of cytokines create damage due to the extreme inflammatory responses they produce, such as high levels of the pro-inflammatory cytokine IL-1β in the lung parenchyma [10] and increased levels of IL-18 in sepsis-induced acute respiratory distress syndrome (ARDS) [11], we propose that inhibiting the caspase-1 protease might decrease the cytokine storm and ameliorate patient healing. Figure 2 shows the potential inhibitors of caspase-1 in this study.
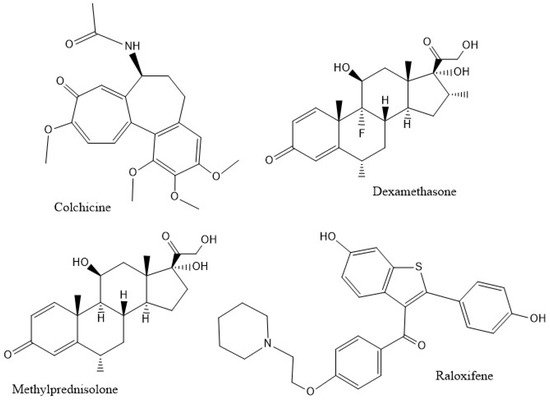
Figure 2. The compounds studied.
Colchicine, an alkaloid isolated from Colchicum autumnale, is among the oldest plant drug compounds known and has been used for centuries to treat gout illness. It inhibits polymerization of microtubules [12]; earlier studies showed that different concentrations of colchicine have diverse effects on the stabilization, dynamics and inhibition of microtubule polymerization [13,14,15]. At low doses (about 10−8 M), the effects are associated with gout inflammation and treatment, and the direct interaction of colchicine with tubulin monomers, or with whole microtubules, leads to protein conformational changes and destabilization. At high doses (about 10−6 M), a second mechanism involves suppression of enzymes that may lead to inflammatory restraint, including caspase-1. Since caspase-1 cleaves pro-IL-1, the release of mature IL-1 decreases upon administration of colchicine, thus inhibiting the propagation of inflammation [16]. Colchicine is used as a treatment for rheumatoid arthritis, a pro-inflammatory cytokine-driven chronic articular condition often accompanied by cardiovascular and lung pathologies. Intriguingly, colchicine shows activity against COVID-19 illness [17], resulting in inhibition of the viral activation of the inflammasome. SARS-CoV-2 drives the assembly of a pro-inflammatory protein complex, the nod-like receptor protein 3 (NLRP3) inflammasome [18], which is composed of the sensor NLRP3, the apoptosis-associated speck-like protein adaptor (ASC or PYCARD) and the effector caspase-1 [19]. Caspase-1 converts pro-interleukin (IL)-1β and pro-IL-18 to their active forms and activates gasdermin-D, permitting large-scale secretion of IL-1β with a subsequent increase in large quantities of pro-inflammatory cytokines [20,21], IL-1β, tumor necrosis factor (TNF) and ligation of Toll-like receptors that activate NF-κB [18] to further upregulate the inflammasome. In SARS-CoV-1, a small envelope protein (E) enhances this reaction that promotes further assembly and activation of the NLRP3 inflammasome [22]. In addition, the creation of IL-1β drives the synthesis of IL-6, a cytokine that induces C-reactive protein (CRP) and which has been identified as a major pro-inflammatory agent in the COVID-19 cytokine storm [23,24,25,26]. Colchicine reduces the concentration of inflammatory biomarkers observed in moderate-to-severe COVID-19 patients and is considered a strong candidate for adjunctive COVID-19 treatment [27]. In eight studies involving 926 COVID-19 patients, 520 patients had standard-of-care therapy while 406 patients also received colchicine and the mortality rate was significantly lower in the colchicine group compared to the control group [28]. Thus, exploring the potential inhibition of caspase-1 by colchicine provides useful information to ameliorate the health condition of COVID-19 patients.
Dexamethasone is a cheap and globally available corticosteroid commonly used as an anti-inflammatory and immunosuppressant. It may modulate inflammation-mediated lung injury and thereby reduce progression to respiratory failure and death. One study showed that, in COVID-19 patients, administration of dexamethasone reduced deaths by one third in patients requiring a ventilator, and one fifth in other patients receiving oxygen only [29]. Dexamethasone also significantly inhibited the activity of the NLRP3 inflammasome and reduced the protein contents of pro-caspase-1, caspase-1 and caspase-1/pro-caspase-1, as well as the cytokines IL-1β, IL-6 and IL-17 in lung tissues. [30]
Since the corticosteroids dexamethasone and methylprednisolone assist in the recovery of severely infected COVID-19 patients needing respiratory support [31], it is of interest to analyze whether the inhibition of caspase-1 is implicated in these cases. Dexamethasone was reported to inhibit LPS-induced inflammation and lung damage by inhibition of the NLRP3 inflammasome and therefore caspase-1 and inflammatory cytokines in mouse macrophages [32]. The subtle structural differences among the three corticosteroids used in COVID-19 treatments (dexamethasone, methylprednisolone and hydrocortisone) compared to inactive anti-inflammatory drugs, have been discussed recently by Draghici and Mor [33]. Comparisons between dexamethasone and methylprednisolone have been carried out by several scholars [34,35,36]. In their paper, Draghici et al. found methylprednisolone more effective than dexamethasone and hydrocortisone, as predicted by in silico evaluation [33]. Other papers have shown the greater effectiveness of methylprednisolone with respect to dexamethasone. In a recent review, an extensive description of clinical trials was discussed, and glucocorticoid action was suggested regarding immunomodulation as a potent COVID-19 pharmacotherapy [37]. As to hydrocortisone, the data reported so far are not clear [38] and will not be further evaluated in the present paper.
The selective estrogen receptor modulator (SERM), raloxifene, has been in use since 1997 for treatment of postmenopausal osteoporosis and breast cancer in the US. Existing clinical studies indicate that estrogens and SERMs, such as raloxifene, modulate immune responses. In particular, raloxifene demonstrates immunoprotective effects [39]. A recent review emphasizes that raloxifene also shows anti-viral activity towards RNA viruses, such as Ebola, influenza A and hepatitis C viruses [40]. Allegretti et al. have performed in vitro experiments that confirm raloxifene to be the most active SERM against SARS-CoV-2 virus at low (micromolar) concentrations [41]. As a result, raloxifene may be usefully repurposed as a drug against COVID-19 [41,42,43]. The in vivo anti-inflammatory effects of raloxifene are described in a study on ovarietomized mice, in which its administration decreased proteinuria-induced renal tubular damage [44]. These mice showed activation of tubular inflammasomes and increased levels of caspase-1-mediated cytokines IL-1β and IL-18. Raloxifene administration resulted in diminished concentrations of these pro-inflammatory cytokines and in less cellular damage.
2. Docking Studies
2.1. Dexamethasone
Docking of dexamethasone on caspase-1 was performed using the PDB 6PZP receptor [48], which consists of two protein subunits containing the inhibitor VX-765. After application of a CHARMm forcefield [49] in Discovery Studio, VX-765 was selected to create a sphere of radius 10 Å. The inhibitor was then eliminated and dexamethasone docked to obtain 15 poses. To perform this docking calculation, the crystal structure atomic coordinates of dexamethasone were used as taken from the CSD Database (refcode DEXMET11) [50].
Examination of the results after docking of dexamethasone at the active site of the caspase-1 receptor reveals some important interactions with Cys285 in the structurally closely related poses 2, 13 and 14. These include a S(Cys285)–C(carbonyl) distance of 3.202 Å (pose 13) (Figure 3 and Figure 4). In addition, the H-bond between F(dexamethasone) and H2N(Arg-B-341), 2.745 Å, provides stabilization of this cysteine–dexamethasone complex, which is absent in pose 2 and pose 14.
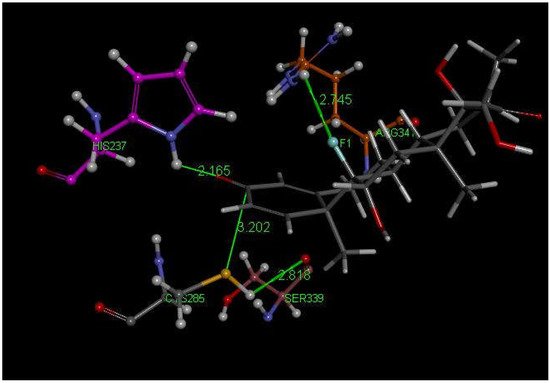
Figure 3. Dexamethasone pose 13 from docking into PDB 6PCP receptor. It has a H-bond between F(dexamethasone) and C(copper)-colored H2N(Arg-B-341), 2.745 Å, which is absent in pose 2 and 14. Purple C-colored His237 has a H-bond to O(carbonyl) of dexamethasone, 2.165 Å. H(Cys285) has a H-bond to polypeptide O(Ser-B-339), 2.818 Å, suggesting potential cleavage of S–H. There is an interaction between S(Cys285) and C(carbonyl) of dexamethasone, 3.202 Å, that is almost perpendicular to the C=O plane.
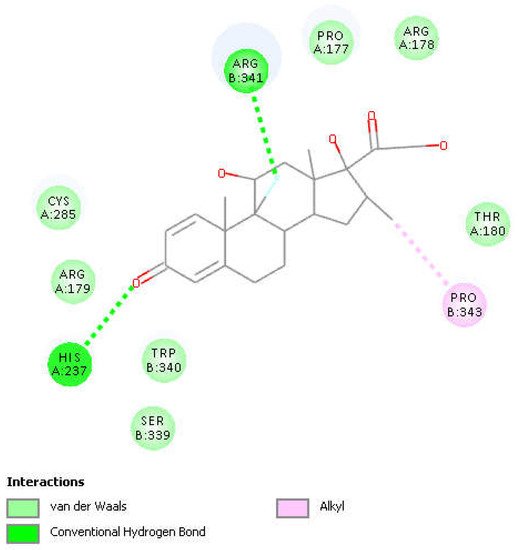
Figure 4. Two-dimensional display of the amino acid interactions of docked dexamethasone pose 13 seen in Figure 3.
The Discovery Studio molecular mechanics program provides the CDocker interaction energy, which is the non-bonded interaction energy (composed of the van der Waals term and the electrostatic term) between the protein and the ligand related to the force field CHARMm. This parameter is −23.9 kcal/mol for dexamethasone pose 13. Figure 5, Figure 6 show HOMO and LUMO of dexamethasone.
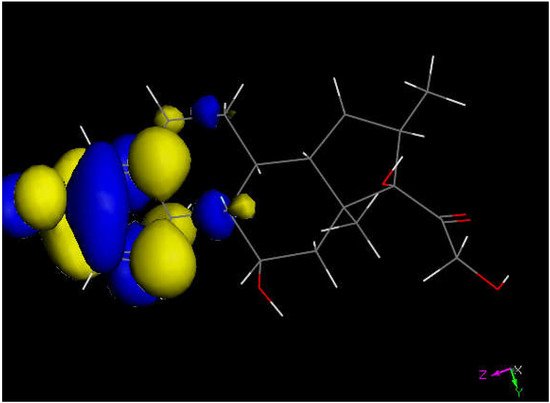
Figure 5. LUMO of dexamethasone suggests the preferential molecular quinone methide moiety (left) prone to reactivity, and a potential addition of a proton can be expected.
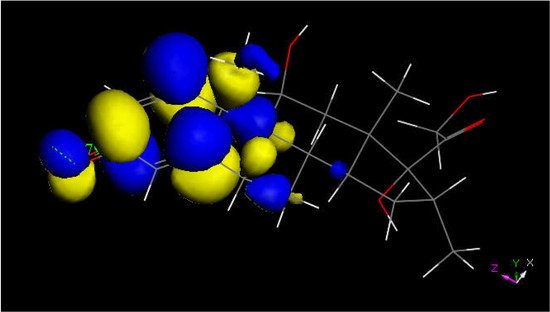
Figure 6. LUMO of protonated dexamethasone also suggests that the quinone methide ring may be prone to reactivity with S(Cys145) thiolate. The [C=OH]+ moiety is located on the left.
2.2. Methylprednisolone
Methylprednisolone is another corticosteroid with anti-inflammatory activity in use for COVID-19 patients [51] and is structurally related to dexamethasone (Figure 2). We used crystal structure atomic coordinates from the CSD database (refcode MTHPRG) [52] for docking methylprednisolone. Pose 8 of this anti-inflammatory drug shows the interaction between S(Cys285) and C(carbonyl) of methylprednisolone equal to 3.135 Å. Further structural details are shown in Figure 7. CDocker interaction energy is −24.0 kcal/mol for pose 8.
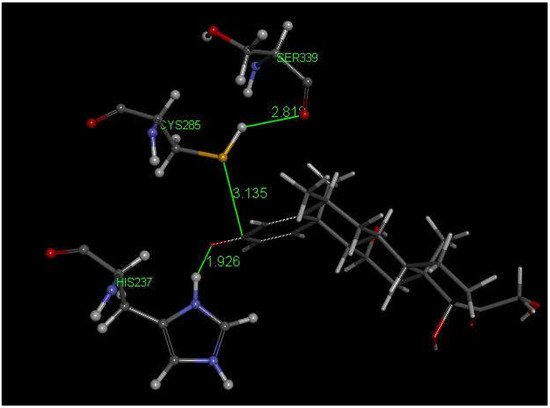
Figure 7. This is pose 8 from docking methylprednisolone into PDB 6PCP receptor. There is an interaction between S(Cys285) and C(carbonyl) of methylprednisolone, 3.135 Å. His237 shows a H-bond to O(carbonyl) of methylprednisolone, 1.926 Å. H(Cys285) has a H-bond to polypeptide O(Ser-B-339), 2.818 Å.
2.3. Colchicine
We next explored colchicine using crystal structure atomic coordinates from the CSD database (refcode COLCDH) for docking [53]. Pose 9 shows the interaction between S(Cys285) and the tropolone C(carbonyl) of colchicine, 3.363 Å, and its CDocker interaction energy is −31.2 kcal/mol (Figure 8 and Figure 9). CDocker interaction energy of pose 1 is −38.5 kcal/mol. Figure 10 and Figure 11 provide HOMO and LUMO of colchicine.
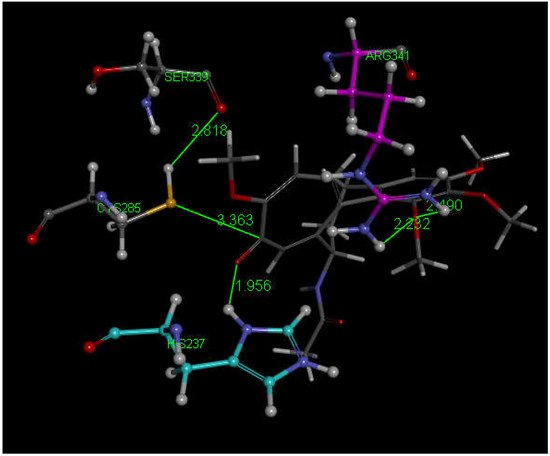
Figure 8. Colchicine pose 9 upon docking into PDB 6PCP receptor. It has H-bonds between one colchicine methoxy moiety and two NH2 groups of purple C-colored Arg-B-341, 2.232 Å and 2.490 Å (center-right). Turquoise C-colored cationic His237 has a H-bond to O(carbonyl) of flat tropolone ring of colchicine, 1.956 Å (bottom). H(Cys285) has a H-bond to O(Ser-B-339), 2.818 Å (center left). S(Cys285) has an interaction of 3.363 Å to C(carbonyl) of flat tropolone ring of colchicine.
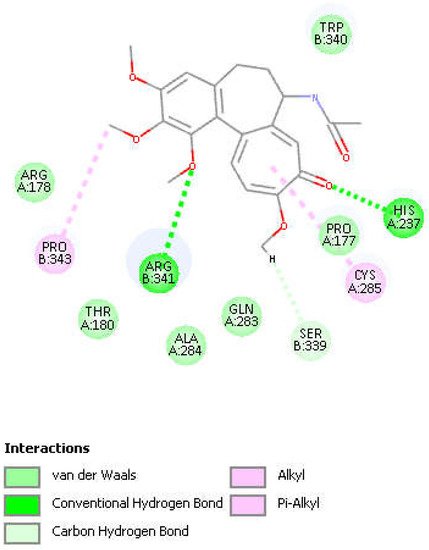
Figure 9. Two-dimensional display of amino acid interactions of docked colchicine pose 9.
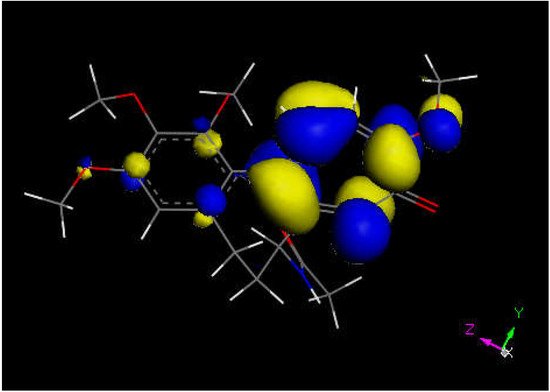
Figure 10. Colchicine LUMO suggests that the aromatic seven-membered ring is more prone for reactivity than the six-membered one.
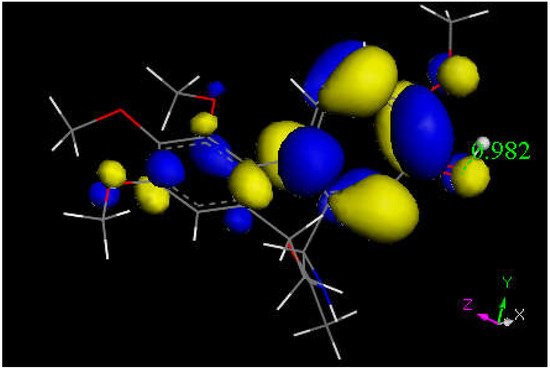
Figure 11. This is the LUMO of protonated colchicine by the tropolone O(carbonyl), showing O-H distance of 0.982 Å. The tropolone ring is more prone to further reactivity than other atoms in this cationic species, suggesting potential interaction with S(Cys145) thiolate. The acetamide carbonyl (bottom) does not appear to be involved in potential reactivity when compared with interaction, though the former tropolone carbonyl is now transformed in the [C=OH]+ moiety (right).
2.4. Raloxifene
Since our docking results repeatedly showed interaction between S(Cys285) and a C(carbonyl), we further enlarged our analysis by investigating also raloxifene, a medicinal compound having anti-inflammatory activity in clinical use against COVID-19 infections which contains a carbonyl functional group. Raloxifene atomic coordinates were taken from CSD (refcode SAQYIR) [54]. Docking of raloxifene into the caspase-1 enzyme (Figure 12) shows pose 2 having an interaction between S(Cys285) and C(carbonyl) of raloxifene of 3.833 Å, whose CDOCKER interaction energy is −39.1 kcal/mol.
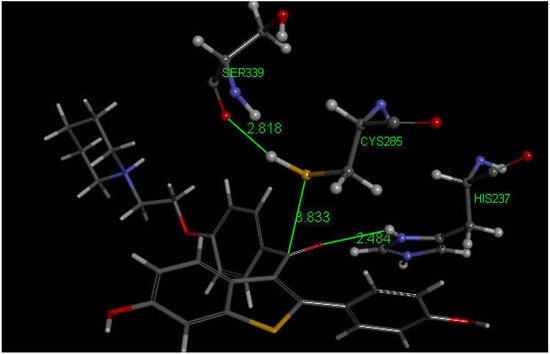
Figure 12. This is pose 2 from docking raloxifene into the PDB 6PCP receptor that shows an interaction between S(Cys285) and C(carbonyl) of raloxifene, 3.833 Å. Cationic His237 has a H-bond to O(carbonyl) of raloxifene, 2.484 Å. Additional interaction between His237 and a raloxifene Ph is seen due to π–π stacking (bottom-right). H(Cys285) has a H-bond to O(Ser-B-339), 2.818 Å. A π–π T-shaped interaction is seen between Trp-B-340 and the nine-membered raloxifene ring (omitted for clarity). Arg-B-341 has a H-bond to the HN by the saturated raloxifene ring.
3. Caspase-1 Inhibitory Mechanism
Overall, these medicinal compounds having anti-inflammatory activity are very promising for treating symptoms of COVID-19 disease, but their chemical mechanism has not yet been clarified. In this study, we take advantage of indications in the literature that inhibition of caspase-1, an enzyme involved in triggering the release of cytokines, may be relevant to the mechanism of anti-COVID-19 therapy. That is, inhibiting caspase-1 may decrease the storm of cytokines involved in response to the SARS-CoV-2 virus attack. The NLRP3 inflammasome promotes inflammation via caspase-1-mediated cleavage and activation of key inflammatory molecules, including active caspase-1, IL-1β and IL-18. It has been demonstrated that the NLRP3 inflammasome is activated in response to SARS-CoV-2 infection and is active in COVID-19 patients [55]. In post-mortem tissues of moderate and severe COVID-19 patients, active NLRP3 inflammasomes have been found upon autopsy. Inflammasome-derived products, such as caspase-1 and IL-18, in sera correlates with the markers of COVID-19 severity, including IL-6. Moreover, higher levels of IL-18 and caspase-1 are associated with disease severity and poor clinical outcome [55]. Since there is strong evidence that production of excessive cytokines creates health problems in COVID-19 patients [55], inhibiting the caspase-1 protease may help patient healing. In fact, the cleaved (active form) of caspase-1 exerts its catalytic activity on the pro-inflammatory cytokines that, after their release, perpetuate the inflammatory response [56]. The docking of four drugs (Figure 1) into the caspase-1 active site highlights promising features and directs us to propose the mechanism shown in Scheme 1 below.
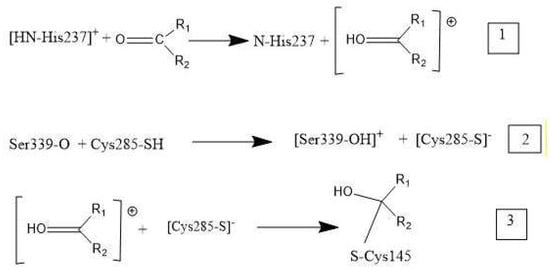
Scheme 1. Mechanism of caspase-1 inhibitors: R1 and R2 are substituents in the four drugs. (1) Cationic [HN-His237] donates a proton to O(carbonyl) of inhibitor. (2) Polypeptide O(Ser339) captures a proton from Cys285, cleaving its S–H bond, and generating S(Cys285) thiolate. (3) The activated carbonyl (step 1) is a nucleophile target for S(thiolate), which generates a covalent S(Cys285)-C(carbonyl) bond.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23031849
This entry is offline, you can click here to edit this entry!