Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Critical Care Medicine
Beyond their role in hemostasis, platelets have emerged as key contributors in the immune response; accordingly, the occurrence of thrombocytopenia during sepsis/septic shock is a well-known risk factor of mortality and a marker of disease severity.
- platelet
- platelet activation
- infection
- critical care
1. Platelets Interactions with Bacteria
The cardiovascular system is usually a sterile environment; however, upon entry to the circulation, bacteria can interact with several cells leading to several complications including bacteremia, sepsis, infective endocarditis, disseminated intravascular coagulation and immune thrombocytopenia purpura. In all these conditions, a common feature is an abnormal platelet function caused by an interaction with the bacteria.
Although the main role of platelets is hemostasis, recently more attention has been focused on the role of platelets in the host response to infection [1,2,3,4,5]. However, under certain circumstances, the platelet response to infection may be a significant part of the problem.
Inflammation and thrombosis both contribute to reducing the spread of pathogens, and platelets migrate to the site of infection and detect pathogens, along with neutrophils [6]; in this manner, platelets migration prevents the dissemination of bacteria already located in the intravascular compartment. In addition to this, platelets use their protrusions at vascular microbreaches to prevent the invasion of extravascular bacteria [7]. This complex defense system contains bacterial spreading and promotes the elimination of bacteria from the circulation by sequestration in the hepatic and pulmonary vasculature. The complement system, which is involved in innate and acquired immune responses to different infections, is the fulcrum of the interplay between inflammation and thrombosis [6]. Some studies identified a shuttling mechanism in the spleen involving complement and platelet and concerning endovascular bacteria which allows to balance rapid clearance of pathogens with the induction of adaptive immune responses [8].
However, even if platelet response to infection is a crucial step in immune response, under certain circumstances it may be a significant part of the problem.
Bacteria can interact with platelets using different mechanisms; they may secrete products (as toxins) that bind to the platelet-causing activation independently of bacterial attachment [9,10] or may bind to platelets. The binding to platelets can be either a direct interaction or an indirect interaction. A direct interaction (Figure 1A) occurs when a bacterial adhesin binds directly to a platelet receptor [11,12]. An indirect interaction (Figure 1B) occurs when a bacterial adhesin binds to a plasma protein (or other soluble elements of the immune system such as immunoglobulins and complement proteins) which bridge the bacteria to a specific receptor on the platelet surface [11,12,13,14,15]. See Table 1 [16,17].
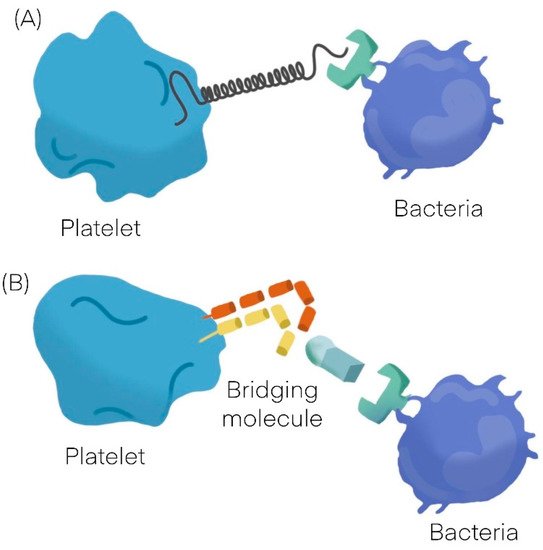
Figure 1. General mechanisms observed in platelet bacterial interaction. (A) Direct interaction. (B) Indirect interaction.
Bacteria can either promote platelet adhesion or can induce platelet aggregation. Platelet adhesion to bacteria is a measure of the strength of the interaction, whereas platelet aggregation is an indication of the quality of the interaction; in contrast to typical platelet aggregation induced by physiological agonists such as adenosine diphosphate (ADP), collagen or thrombin, bacteria induce an all or nothing response. In other words, there is a threshold concentration of bacteria, above which there is maximum aggregation and below which there is no aggregation at all [16,17]. Another specific feature of platelet aggregation induced by bacteria is “lag time,” which is a distinct pause in time before aggregation takes place. When a soluble agonist such as ADP is added to a suspension of platelets, the aggregation response occurs within a few seconds; otherwise, when bacteria are added to a platelet suspension, there is a concentration-dependent delay in the aggregation response [18]. Depending on bacteria, platelet aggregation may be preceded by a short lag time of around 2–5 min [11,19] or by a long lag time of about 12–18 min [13,20]. A short lag time usually indicates a direct interaction between the bacteria and the platelet, whereas a long lag time generally indicates an indirect interaction. The length of time relates to how long it takes for the bacteria to bind the bridging molecule and cross-react with the reciprocal receptor on the platelet [16].
The activation of platelets by bacteria can lead to different specific problems. If activation occurs in a localized manner, it can lead to thrombus formation; instead, a more systemic activation can lead to platelet consumption. Finally, activated platelets secrete many cytokines and other mediators that can trigger pathological processes. Infective endocarditis (typically due to Staph. aureus or an oral Streptococcus), in which a bacteria–platelet thrombus develops on the valve, is a typical example of a thrombotic complication of bacterial infection and can either lead to valve failure or the formation of a septic embolus [21]. During septicemia, platelet activation by systemic bacterial infection may lead to thrombocytopenia and bleeding complications due to platelet sequestration [22,23]; and this outcome relates to the entity of thrombocytopenia [24,25].
When activated, platelets secrete their granule contents, which contain at least 300 different proteins including cytokines and vascular activating factors [26,27]. These cytokines play a key role in the pathogenesis of atherosclerosis [26,28,29,30,31] and may also explain the association between infection and cardiovascular disease. As well as causing thrombocytopenia, sepsis also leads to shock due to endothelial inflammation and subsequent vascular leakage. Activated platelets play a key role in mediating endothelial damage [23,32,33].
Table 1. Bacterial–platelet interaction can vary depending on etiologic agent.
| Platelet-Bacteria Interactions | ||
|---|---|---|
| Direct adhesion | Indirect adhesion | |
| Short lag | Bridging protein | |
| S. sanguinis [11] | S. aureus [12,13,14] | Fibrinogen |
| S. aureus [12,13,14] | H. pylori [19] | Fibronectin |
| VWF | ||
| Direct adhesion | ||
| Long lag | ||
| S. gordonii [20] | ||
| S. sanguinis [11] | ||
| Non aggregating | ||
| S. gordonii [20] | H. pylori [19] |
2. Platelets Interactions with Viruses
The relationship between activated platelets and immune response is maintained even when the infectious microorganisms are viruses [34,35,36,37]. Viral infection of cells begins with virus binding to a surface receptor that mediates its internalization, and platelets express various pattern recognition receptors (PRR) able to mediate binding and entry of various viruses [36,38,39,40]. The immune response against viruses is supported by consequent platelet activation and manifests itself through different mechanisms, such as the release of chemokines that promote endothelial signaling and leukocyte migration or by physically interacting with leukocytes [41,42,43]. Moreover, while traditional platelet activation by G-protein-coupled receptors is usually rapid, platelet PRR activation in responses to infections and immune stimuli can be delayed and sustained, persisting hours after initial aggregation and secretion [34,35,36,37,38,39,40,41,42,43,44].
Resultant virus-mediated thrombocytopenia is generally multifactorial; viruses use different strategies to decrease the levels of circulating platelets, either by decreasing platelet production or increasing platelet destruction. Platelets are produced in the bone marrow by megakaryocytes, and viruses can interfere with platelet production at various steps of development [36]. Some viruses, such as simian immunodeficiency virus (SIV) and human herpes virus, can influence the cytokine profile of the host, resulting in altered thrombopoietin (TPO) production in the liver [45,46,47]. Some others, including hepatitis C virus (HCV), also directly interfere with TPO production by destruction of liver tissue [48]. Moreover, human immunodeficiency virus (HIV), cytomegalovirus (CMV) and HCV replicate in megakaryocytes modulating their proliferation and function [49,50,51,52]. Nevertheless, thrombocytopenia induced by decreased platelet production is observed at later stages of infection; otherwise, rapidly induced thrombocytopenia during viral infections is mediated via enhanced platelet destruction. The most rapid way of platelet destruction occurs via direct interaction between platelets and viruses through a variety of receptors and it is mainly mediated by surface lectins, integrins and TLR [52,53]. Rotavirus utilizes the collagen receptor GPIa/IIa to bind to platelets [54,55], while hantavirus and adenoviruses interact with platelets via the fibrinogen receptor GPIIb/IIIa, the most abundant platelet integrin. [56] The Epstein–Barr virus’ (EBV) interaction with platelets occurs via complement receptor 2 (CR2) [57]; HIV and dengue virus activate platelets by binding to lectin receptors and a cell-specific intercellular adhesion molecule [58]. These direct interactions often result in platelet activation and adhesion of activated platelets to leukocytes. Platelet binding to neutrophils leads to phagocytosis of platelets and platelet activation itself stimulates platelet clearance in the liver and spleen [59,60]. However, platelets are not only activated by direct interactions with viruses. Host defense mechanisms in response to viral infections can also lead to platelet activation and decreases platelet life span [61]. Moreover, the B-lymphocyte production of antibodies against some viruses also interferes with platelet survival. These antibodies, which usually target surface glycoproteins of viruses, shows cross-reactivity with platelet surface integrins such as GPIIb/IIIa. That is called idiopathic thrombocytopenic purpura (ITP) or platelet autoantibody-induced thrombocytopenia and it has been described for HCV, HIV, CMV, EBV, hantavirus, varicella zoster virus, herpes viruses, and severe acute respiratory syndrome coronavirus [62]. The interaction between platelets and viruses is crucial even in the pathogenesis of the COVID-19 syndrome. Platelets from COVID-19 patients are more activated, aggregated faster and have increased expression of monocyte tissue factor [39,40]. Functional assays showed that platelets from COVID-19 patients are more responsive, sensitized to release inflammatory cytokines and adhere more efficiently [33]. All things considered, the data suggest that platelets may have the potential to contribute to the thrombo-inflammation in COVID-19 [33].
3. Cellular Changes in Platelet Structure and Function during Infection
The interplay between platelets and bacteria or viruses reported above can affect the structure and the function of platelets in several ways. The most known effect of infection is a fall in platelet count, which may show different levels of severity depending on the lowest level of platelet reached. The occurrence of thrombocytopenia has long been recognized as an independent risk factor for worst outcomes during infection, and the degree of thrombocytopenia is used as a marker of the severity during sepsis [33,63]. Indeed, the platelet count is included in the Sequential organ failure assessment (SOFA) score [64], the alteration of which is crucial in the diagnosis of sepsis.
Beyond the well-established prognostic finding of thrombocytopenia, there are other less-known platelet characteristics that may be evaluated during infection. Two indices of platelet morphology are easily available in most laboratories and have been shown to be affected by concurrence infections: the platelets distribution width (PDW) and the mean platelet volume (MPV). The PDW is a parameter of platelet heterogeneity, while MPV is a measurement of the average size of platelets. Platelet size is usually between 1.5 and 3 μm. Large platelets (3–7 μm) are called macrothrombocytes, whereas platelets reaching the size of erythrocytes or lymphocytes (larger than 7, up to 20 μm) are designated giant platelets [65]. Healthy subjects usually have less than 5% of large platelets, but infection-induced platelet activation is associated with major shape change due to cytoskeletal changes, including filopodial and lamellopodial extensions. These changes affect platelet size and variability and, therefore, MPV and PDW have recently been suggested as markers of platelet activation [66,67,68,69]. High PDW and MPV values were associated with 90-day mortality in patients with septic shock [62,63,64,65]; this was also in experimental animal models of endotoxemia [69]. Notably, studies conducted in non-infected critically ill patients, such as cardiac arrest [70], have not found any prognostic role of these morphological indices; these results suggest a direct role of infection on platelet morphologies, and a recent study confirmed this hypothesis [68]. As a result, both PDW and MPV can be used as a marker of infection severity and they are independent predictors of mortality during infections.
Outside the platelet morphological changes, several other biological changes can be described after platelet activation, which include the expression of different platelet receptors. P-selectin, which can bind leukocytes, is only expressed on the surface of activated platelets [71] and can be responsible for neutrophil-platelet aggregates in the circulating blood [72]. Similarly, CD40-Ligand (CD40L) is expressed on activated platelets, and can trigger an inflammatory response by interacting with CDBoth P-selectin and CD40L can be measured with a bead-based multiplex immunoassay and were associated with worse outcomes both in bacterial [73] and viral infections [74]. Remarkably, experimental studies showed compromised host defense to infection in P-selectin-deficient mice [75]; it appears that only a dysregulated platelet activation can be deleterious in patients with infections.
This entry is adapted from the peer-reviewed paper 10.3390/cells11030424
This entry is offline, you can click here to edit this entry!