1. The Complement System (CS)
The complement system (CS) plays a key role in the defense against pathogens as part of the human immune system [
1,
2]. It consists of more than 30 soluble proteins synthesized mainly by the liver (but also by leukocytes), which are present in the plasma in an inactive form [
3], as well as membrane-bound regulators and receptors that interact with various cells and mediators of the immune system [
4]. Activation of the complement system involves a cascade of enzymatic and non-enzymatic reactions that culminate in opsonization by various opsonins (e.g., C3b and C4b) of the pathogens or pathogen-virus-infected cells and then lysis of these cells by a set of proteins that form a membrane attack complex (MAC) [
5,
6]. In addition, activation of the complement system leads to the production of anaphylatoxins—potent proinflammatory molecules. Complement also served to mediate clearance of immune complexes and damaged self cells or cell debris and to mediate phagocytosis by neutrophils and monocytes [
7,
8]. A limited number of reports indicate that complement may contribute to the regulation of the anti-inflammatory response. This process involves T lymphocytes (Treg), crucial in the production of anti-inflammatory cytokines such as transforming growth factor-β (TGF-β), IL-10, and IL-35 [
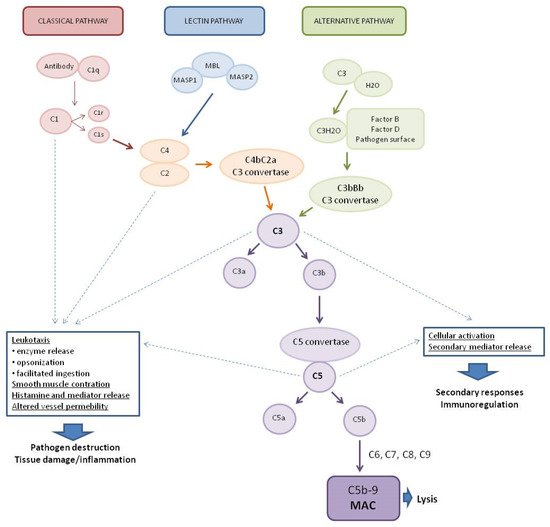
8]. The system is activated by three main pathways: the classical pathway (CP), the lectin pathway (LP), and the alternative pathway (AP) (
Figure 1) [
9].
Figure 1. Pathways of complement system activation. MASP1—mannan-binding lectin associated serine protease-1, MASP2—mannan-binding lectin associated serine protease-2, MBL—mannose-binding lectin, and MAC—membrane attack complex.
The classical complement system pathway is referred to as “antibody-dependent” because of the involvement of IgM/IgG antibodies in the activation. Protein C1q (part of the C1 complex which consists of six molecules of C1q, two molecules of C1r, and two molecules of C1s) binds to the Fc region of complement-fixing antibodies (generally IgG1 and IgM) attached to pathogenic surfaces and pathogen-infected cells. This results in the activation of C1r and C1s proteases in the C1 complex [
10,
11]. C1s cleaves C4 and C2 proteases into large fragments (C4b, C2a) and small fragments (C4a, C2b). The larger fragments combine to form the C4bC2a complex on the pathogen surface which leads to the cleavage of C3 into anaphylatoxin C3a and opsonin C3b. The production of C3 convertase is the point where three complement pathways converge and afterwards have common steps to form MAC [
1]. The lectin complement system pathway is similar to the classical pathway, but is independent of immunoglobulins. It does not recognize antigen antibody complexes but employs germline-encoded pattern-recognition receptors (PRRs) such as mannose-binding lectin (MBL) and ficolins [
5]. When MBL recognizes and binds to carbohydrates in pathogen-associated molecular patterns (PAMPs), such as those found in viruses [
12], MBL-associated serine proteases (MASPs, MBL-associated serine protease-2 (MASP-2), and MBL-associated serine protease-1 (MASP-1)) are activated to cleave complement components C2 and C4, which then leads to the generation of C3 convertase [
13,
14,
15]. The alternative complement system pathway, in contrast to the classical and lectin pathways, consists of three processes that partially overlap. In the alternative pathway, activators include viruses [
16]. Activation of the C3 convertase in this pathway occurs slowly in plasma and lead to the formation of C3H2O. C3 activation is induced by the presence of various surfaces which lack complement regulatory proteins which adsorb C3 to the surface to induce its conformational changes. C3H2O can then bind factor B (FB) to induce another conformational change as FB is cleaved into two components (FBb and FBa) by factor D (FD) [
5]. This convertase begins to cleave C3 into C3a and C3b, in a manner analogous to the C4bC2a convertase in the classical and lectin pathway. The resulting C3b can bind to cell surfaces and FB to form the predominant convertase in the alternative pathway, i.e., C3bBb [
2,
17]. This C3bBb complex can be further stabilized by properdin, protecting it from the inactivating factor H (FH) and factor I (FI) [
18]. All surface-bound C3 convertases, regardless of their origin, can induce an amplification branch of the alternative complement system pathway through the activation of C3 [
19], which increases the density of deposited C3b and gradually leads to the formation of convertases that contain an additional C3b molecule (C4b2b3b or C3bBb3b) shifting its substrate specificity towards C5. The C5 convertase of the alternative pathway cleaves C5 into the anaphylatoxin C5a and the C5b fragment. When C5b binds to C6 and C7, the complex is inserted into cell membranes and interacts with C8, inducing the binding of several C9 units to form the MAC complex C5b6789 [
20].
Activation of the complement system, regardless of the pathway, results in the generation of three broad effector pathways that enable the complement system to perform its physiological functions in host defense: direct lysis of target surfaces by the MAC, alerting and stimulating the immune system by producing potent proinflammatory anaphylatoxins, and opsonization of target surfaces by opsonins C4b, C3b, and C3bi [
2,
5,
17]. MAC formation and targeted lysis are important effectors of the complement system’s anti-pathogenic actions [
9]. Some cleavage products and the complement system activation products can act as anaphylatoxins and have broader immune regulatory functions. Most notably, the cleavage products C3a and C5a can be generated by all three pathways and can act as potent immune regulators, whereas C4a is generated exclusively by the classical and lectin pathways [
21]. C3a and C5a can affect chemotaxis of eosinophilia, fibroblasts, macrophages, mast cells, and monocytes to the site of infection and inflammation, C5a alone is responsible for neutrophil recruitment [
6], while C4a acts as an effector protein and increases endothelial cell permeability and enhances stress fiber formation [
22]. In addition to their roles in chemotaxis, C3a and C5a have been implicated in the regulation of vasodilation, increased vascular permeability [
23], and the production of various cytokines, including IL-1β, IL-8/CXCL-8, CCL5, IL-6, and tumor necrosis factor-α (TNF-α) [
24,
25].
The complement system, with its ability to form channels in the cell membrane, induce phagocytosis, and cause mast cell degranulation, is a dangerous weapon, and without constant supervision by the various mechanisms regulating its activity (Table 1), it could easily lead to cell and tissue damage in our body.
Table 1. Factors regulating the activity of the complement.
2. The Complement System at the Crossroads of the Innate and Adaptive Immune Response
The complement system is a part of the immune system and plays the role of a functional bridge between innate and adaptive immune responses that allows an integrated host defense against pathogens [
9]. The humoral immune response is designed to protect extracellular spaces by activating effector and memory B cells and producing antibodies, leading to the neutralization and opsonization of the pathogen and providing immune memory against reinfection [
5]. Complement system effectors are involved in humoral responses at multiple stages of B lymphocyte differentiation [
35]. The complement system enhances B cell immunity mainly through complement receptors (CR), complement receptor type 1 (CR1) (CD35), and complement receptor type 1 (CR2) (CD21) expressed on B lymphocytes and follicular dendritic cells (FDCs). The CR2 receptor forms a receptor complex with the signaling protein CD19 and protein CD81 to form a receptor complex (CD21-CD19-CD81) of B lymphocytes. This supports enhanced B cell receptor (BCR e.g., surface immunoglobulins)-mediated signaling upon encountering a pathogen coated with complement system opsonins, resulting in a lowered threshold for B lymphocyte activation [
36,
37]. Coupling C3d to a low-affinity antigen which in the absence of coupling would cause B cell death, results not only in survival but also in B cell activation and antibody production [
38]. Furthermore, the CR2 receptor mediates antigen-independent signals that are essential for B cell survival [
39].
A regulatory effect on B lymphocytes is also induced by anaphylatoxins. Anaphylatoxin C3a causes the suppression of polyclonal B lymphocyte responses, while C5a promotes naive B cell migration and memory [
40,
41]. The complement system also influences T cell-associated responses. Experiments on mice lacking complement system inhibitory proteins (Decay-accelerating Factor (DAF) and CD59) highlight the important regulatory role of the complement system in the development of T cell immunity [
9,
42]. DAF deficiency increases cytokine production by T cells, and CD59 ligation decreases CD4+ T cell activation [
43]. The interaction between antigen-presenting cells (APC) and T lymphocytes induces the local production of C3, C5, as well as FB and FD. Moreover, C3aR and C5aR receptors are upregulated on T lymphocytes, whereas DAF production is downregulated. Local production of complement system components from immune cells allows signals to be transduced by C3aR and C5aR receptors in an autocrine and paracrine manner. Complement system component C3 activated in the alternative pathway can increase the production of proinflammatory cytokines from T cells [
44,
45]. Induction of Th1 responses also depends on the activation of C3aR and CD46 receptor on T cells through their T cell-derived ligands [
46]. In contrast, the absence of C3aR and C5aR receptors leads to reduced complement protein and receptor regulation, lack of expression of co-stimulatory molecules, impaired production of cytokines (IL-1, IL-23, and IL-12), induction of the Treg cell response, and inhibition of T cell proliferation [
47,
48,
49].
3. Antiviral Activity of the Complement System and Viral Strategies for Reducing the Complement System Action
All three complement system pathways can lead to viral opsonization and deposition of complement system components upon activation. The outcome of this response is highly dependent on the infectious agent and can enhance viral infection, suppress viral infection, or be dysregulated by the expression of certain viral proteins [
50]. The MBL protein of the lectin pathway can interact with numerous viral antigens and have different effects on neutralization or increased viral replication. MBL can directly bind the glycoprotein (GP) of the Ebola virus (EBOV) [
51]. High doses of MBL, relative to other complement proteins, can enhance infection with nonreplicating (pseudotyped) EBOV-GP virus in primary human macrophages and human monocyte-derived macrophage cell lines [
51]. This may be due to the activation by viral ligands of the C1QBP (gC1qR) translocation, which inhibits RIG-1 and then inhibits antiviral signaling by downregulating type I interferons. It is speculated that EBOV-MBL complexes activate C1QBP, which then negatively regulates RIG-1 inhibition of viral infection, thereby enhancing viral proliferation [
51]. Additionally, MBL opsonization of the EBOV GP prevents GP binding to DC-SIGN (dendritic cell-specific intercellular adhesion molecule) and, therefore, neutralizes EBOV (pseudotype) [
52]. Thus, in the context of EBOV infection, MBL effects appear to be dependent on the cellular target and the relative concentrations of other complement system protein components. It has also been shown that in vitro infection by the human immunodeficiency virus (HIV) of CD4+ H9 lymphoblasts is inhibited by MBL from human serum. In addition, MBL is able to selectively bind to HIV-infected H9 cells and HIV-infected U937 cell line [
53]. These results indicate that MBL inhibits viral entry to susceptible cells. Similarly, in another study [
53], gp120 HIV bound directly to MBL. In a later study about HIV, MBL was shown to be sufficient for virus opsonization but not neutralization [
54]. It also showed that both the primary isolates (PI) of HIV and cell line-adapted HIV, despite binding to MBL, are relatively resistant to neutralization by MBL at the levels normally present in the serum. However, binding and opsonization of HIV by MBL may alter virus trafficking and viral-antigen presentation during HIV infection [
54]. In another study, ref. [
55] complement opsonization of herpes simplex virus-2 (HSV-2) both by human serum and by seminal plasma, produced enhanced infection of DCs and resulted in greater productive infection compared to free, nonopsonized HSV-2. Furthermore, opsonization gave rise to significantly higher gene expression of all inflammatory (TNF-α, IL-6, IL-1β) and antiviral factors (IFN-α, IFN-β, MX1), but at the protein level these differences between free and complement-opsonized HSV-2 were not as clear as at the gene level. The enhanced infection induced by complement-opsonized virions required the functional complement receptor 3 (CR3). In contrast, the presence of complement in combination with HSV-1 or HSV-2-specific antibodies decreased infection, inflammation, and antiviral responses of DCs. HSV-2 infection of DCs required endocytosis and endosomal acidification, as inhibition of these cellular events decreased infection [
55].
Other complement proteins and subsequently the complement system activation products can opsonize virus particles. In the case of dengue virus (DENV) and West Nile virus (WNV), viral neutralization occurs in a C3- and C4-dependent manner after MBL binding. In the case of WNV, neutralization was achieved despite reduced levels of C5, indicating that neutralization did not require MAC generation [
56]. In the case of monkey simian virus 5 (SV5), complement-mediated neutralization is achieved mainly through C3 deposition and the formation of viral aggregates rather than viral lysis [
57]. Similarly, the complement activation in the presence of Influenza A Virus (IVA) results in viral aggregation and opsonization of the hemagglutinin receptor, although IgM antibodies and activation of the classical pathway are required to achieve neutralization [
58]. Some complement proteins may also have a protective intracellular function. Intracellular C3 signaling induces the production of proinflammatory cytokines (IFN-β, IL-6, and IL-1β) by nuclear factor kappa B (NF-κB), and activates interferon regulatory factor (IRF), and activator protein-1 (AP-1). Detection of intracellular C3 has been shown to be dependent on mitochondrial antiviral signaling protein (MAVS) and independent of PAMPs and pattern recognition receptors (PRRs) [
59]. Infected host cells that present viral antigens on the cell surface membrane can activate the classical pathway as the antigens bind IgM/IgG, and induce complement-dependent cytotoxicity (CDC). The infected cell is then lysed by MAC to reduce the virus titer. However, some viruses have evolved self-defense mechanisms against the action of the complement system to enable their survival [
50] (
Table 2).