Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Somatostatin (SST) is a small peptide that exerts inhibitory effects on a wide range of neuroendocrine cells. Due to the fact that somatostatin regulates cell growth and hormone secretion, somatostatin receptors (SSTRs) have become valuable targets for the treatment of different types of neuroendocrine tumours (NETs).
- neuroendocrine tumour
- somatostatin
- somatostatin receptor
- somatostatin analogue
- PRRT
1. Introduction
Neuroendocrine tumours (NETs) represent a heterogeneous group of neoplasms and can originate from neuroendocrine cells in any organ of the body [1]. For all NETs, overall survival rates are approximately 55% and 45% five and ten years after diagnosis, respectively [2]. The classification and clinical picture of each NET differs based on the organ of origin; however, all NETs share the expression of somatostatin receptors (SSTRs), which have become valuable targets for somatostatin analogue (SSA) therapy. Symptom management is the most prevailing therapy in patients with functioning NETs.
2. Somatostatin Signalling
Somatostatin (SST), also known as the somatotropin release-inhibiting factor (SRIF) or growth hormone-inhibiting hormone (GHIH), is a cyclic peptide that exerts inhibitory effects on the endocrine and exocrine hormone secretion [3][4],. The growth hormone (GH), prolactin (PRL), thyrotropin (TSH), cholecystokinin, gastric inhibitory peptide, neurotensin, motilin, gastrin, secretin, glucagon, insulin, pancreatic polypeptide, and cytokines in immune cells are all inhibited by SST [3][5]. The effects of SST on exocrine hormones include the suppression of amylase secretion from salivary glands; the inhibition of hydrochloric acid, pepsinogen, and intrinsic factors in the gastrointestinal mucosa; the reduced secretion of pancreatic enzymes and bicarbonate, and the reduced secretion of bile from the liver. SST also prevents the absorption of glucose, fat, and amino acids, helps to regulate gastrointestinal motility by delaying late gastric emptying, weakens gallbladder contractions, and lengthens small intestinal transit time. SST also reduces the time between migrating motor complexes and accelerates early stomach emptying. Immunoglobulin production and lymphocyte proliferation have both been found to be inhibited by SST in lymphoid tissues [5].
SST is produced in many locations of the body, primarily in the pancreas, gastrointestinal (GI) tract, central nervous system (CNS), and hypothalamus [6]. Both isoforms of SST (SST-14 and SST-28) are derived from a 116-amino acid precursor protein, known as pre-prosomatostatin, which is cleaved into 92-amino acid prosomatostatin. To generate SST-14 and SST-28, prosomatostatin undergoes C-terminal post-translational processing, which results in the production of a predominant 14-amino acid molecule as well as a larger, N-terminally extended 28-amino acid form (Figure 1) [7][8]. While the shortest isoform is secreted mainly from the β-cells of the pancreas, the SST-28 is the product of GI cells [9][10]. SST-14 has wide-ranging effects, including the inhibition of GH, TSH, and corticotropin (ACTH) within the pituitary, as well as the inhibition of glucagon and insulin in the pancreas [11]. SST-28 refers to the endogenous pro-form of SST, which regulates the inhibition of the hormones previously mentioned in the context of SST-14 [12]. SST-28 is known to be more potent than SST-14 with respect to its effect on GH, PRL, insulin, glucagon, TSH, and gonadotropins (LH and FSH) secretion [13]. It is estimated that 65% of the circulating SST is produced and secreted by the D-cells of the GI tract, 30% is produced by the CNS (hypothalamus and amygdala), and the remaining 5% is produced by pancreatic β-cells [7][14]. Both SST active forms are stored in secretory granules and have a short (~1 min) bioactive half-life time (t½) once released into the circulation [3][10].
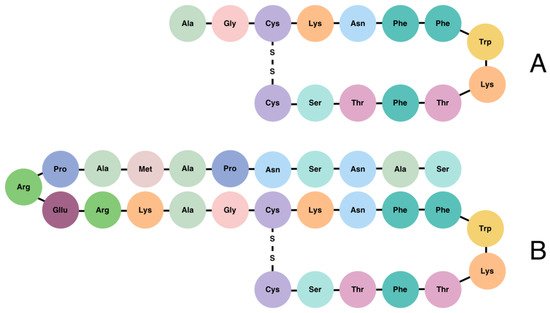
Figure 1. Structure of somatostatin-14 (A) and somatostatin-28 (B).
The activity of SST is mediated by its binding to five subtypes of SSTRs, each encoded by five different genes segregated on chromosomes 14, 17, 22, 20, and 16, respectively [15]. The SST receptor subtypes (SSTR1 through SSTR5) share signalling pathways and structural features [16]. Two isoforms of SSTR2 exist (SSTR2A and SSTR2B), and are produced via alternative splicing [17][18]. These variants of SSTR2 differ in the length of their C-terminal cytoplasmic tails as well as their ability to couple to adenylyl cyclase; as a result, SSTR2A and SSTR2B may activate alternative signal transduction pathways [19][20]. SSTRs belong to the superfamily of G-protein-coupled receptors (GPCRs) and play a crucial role in vertebrate development, metabolism, and growth [21]. GPCRs represent the largest family of human membrane proteins, characterized by a core of seven transmembrane helices that are connected by three extracellular loops (ECLs) and an amino terminus [15][22]. The activation of SSTRs usually results in the inhibition of adenylyl cyclase and the reduction in intracellular Ca2+, which result in the inhibition of cell proliferation and the secretion of signalling molecules [23][24]. Firstly, SST activates the SSTR which interacts with the G protein, consisting of α-, β-, and γ-subunits, thereby modulating several downstream second messenger systems. The α-subunit reduces the affinity for guanosine diphosphate (GDP), and because the concentration of guanosine triphosphate (GTP) is higher in the cytoplasm, GDP is, thus, replaced by GTP. Thereafter, the Gα protein dissociates from the receptor and subunit complex and modulates the activity of several intracellular pathways [15].
SSTRs are expressed throughout many tissues of the body, including the hypothalamus and pituitary, GI tract peripheral organs, and pancreas, as well as kidneys, thyroid, lungs, and immune cells. Moreover, SSTR expression has been reported in numerous types of tumours [15]. In the GI tract, SST regulates the release of gastric acid by a negative feedback mechanism of paracrine effects. The feedback pathway involves stomach D-cell SST release in response to direct stimulation by gastrin, and this indirectly inhibits further gastric release from G-cells [25]. Within the hypothalamus, SST indicates the inhibition of GH, LH, and TSH release from the pituitary [26]. SST binds to the β-cells of the pancreas to inhibit voltage-gated calcium channels, resulting in the suppression of the early insulin response to glucose and, thus, downmodulating the storage of energy in adipose tissue [27]. SST suppresses several immune functions, such as lymphocyte proliferation, immunoglobulin production, and the release of proinflammatory cytokines such as interferon-γ (IFNγ) and interleukin-8 (IL-8) [28][29]. The effect of SST under physiological conditions is partially determined by the types of SSTRs expressed on the tissue’s surface[6]. For instance, SSTR2 and SSTR5 have been reported as the most abundantly expressed receptors. Both show inhibitory effects on GH and ACTH within the pituitary gland, on insulin, within the β-cells of the pancreas, and on glucagon-like peptide 1 (GLP-1), IFN-γ, and reduce the secretion of gastric acid [16][24][30]. SSTR2 is extensively expressed in pulmonary endocrine tumours, including typical and atypical carcinoids and non-endocrine lung cancers such as adenocarcinoma and small cell lung cancer [31]. Receptor expression profiles differ between patients and even between tumours within the same patient. SSTR2 is expressed in about 80% of GI tract and pancreatic endocrine tumours according to Reubi and colleagues [32]. Among the tumours, SSTR2A is the most commonly expressed receptor subtype. The expression of SSTR2A has been reported in gastrinomas, insulinomas, gliomas, medulloblastomas, paragangliomas, and neuroblastomas [33][34]. Neuroblastomas are the most common malignancy among children. These types of tumours are typically associated with a high expression of SSTR1 and SSTR2, which usually indicates a good prognosis for patients [35]. SSTR2A correlates with the overall survival rate in patients with medullary thyroid carcinoma and is considered as a favourable prognostic marker in stage IV patients [36]. The activation of SSTR1 shows antisecretory effects on the GH, PRL, and calcitonin, whereas SSTR3 regulates antiproliferative signalling and induces apoptosis in several cell types [30][37]. The role of SSTR4 remains mostly unknown, but it may be linked to the inflammation of the intestine. SSTR4 has been identified as a key player in the inflammatory effects exerted by SST, either through the direct targeting of inflammatory cells or via the indirect modification of cells that synthesize and release pro-inflammatory mediators [38]. Pro-inflammatory mediators are released from capsaicin-sensitive sensory nerve endings during inflammation. These mediators primarily include tachykinins (substance P and neurokinin A) and the calcitonin gene-related peptide, which may be involved in the sympathetic reflex inhibition of GI propulsion, ultimately initiating an inflammatory cascade [38][39]. The expression of SSTR4 has also been detected in the lungs, heart, and placenta [16].
Expression levels of SSTRs have been reported in the majority of NETs and non-neuroendocrine tumour types, including pancreatic NETs (PanNETs), pituitary NETs (PitNETs), and gastroenteropancreatic NETs (GEP-NETs), as well as hepatocellular carcinoma and breast cancer [23][40][41][42]. Since SSTRs are located on the surface of tumour cells, they have the potential to serve as diagnostic markers and be used for SSA treatment strategies [38].
3. Neuroendocrine Tumours
NETs are a heterogeneous group of generally slow-growing neoplasms of epithelial origin with variable clinical prognoses and behaviour [43][44]. NETs, not to be confused with neuroendocrine carcinomas (NECs), are believed to originate from hormonally programmed neuroendocrine precursor cells that undergo tumourigenic mutational events. For this reason, NETs mostly consist of well-differentiated neuroendocrine cells. Normally, neuroendocrine cells can be either diffusely distributed in the mucosal membrane, as in the case in the digestive system, or they can form organised cell clusters or organs of endocrine function, such as pancreatic islets or the pituitary gland [1]. Neuroendocrine cells are widely distributed in the human body and, for this reason, NETs can occur in virtually any location. However, NETs occur most commonly in the GI tract, pancreas, and lungs [45].
NETs are generally subdivided by their proliferative activity using the mitotic and/or Ki67 index. G1 NETs are classified by <2 mitoses/10 high-power fields and a Ki-67 index of <3%, G2 NETs are classified by 2–20 mitoses/10 high-power fields or a Ki-67 index of 3–20%. Well-differentiated G3 NETs are classified by >20 mitoses/10 high-power fields or a Ki-67 index of >20%, and poorly differentiated G3 NECs are classified by >20 mitoses/10 high-power fields or Ki-67 and expression alterations of p53 and Rb1 [1][46][47][48]. Further classification depends on tumour location and functionality, and it is not uniform across different centres.
Due to their hormonal origin, NETs can synthesise and secrete cell-type-specific peptide hormones and neuroamines, and, therefore, are characterised as functioning NETs [49]. For example, PitNETs can additionally secrete GH, PRL, or other pituitary hormones, thereby elevating hormone concentrations in the circulation, leading to hormonal dysregulations and characteristic clinical syndromes [50][51]. About 60–90% of PanNETs are non-functioning and do not show significant symptoms. Functioning PanNETs are uncommon and are usually associated with the increased secretion of various hormones, including insulin, gastrin, ghrelin, vasoactive intestinal peptide (VIP), glucagon, and SST [52]. Most GEP-NETs are non-functioning and present moderately late; in turn, functioning tumours cause distinct clinical syndromes resulting from the production of various bioactive peptides or amines. For instance, active gastric NETs (GNETs) are known to secrete histamine, yet the duodenum produces secretin, gastrin, gastric inhibitory polypeptide, and motilin [53]. Furthermore, NETs have the capacity to modify secreted hormones and peptides at the genetic level. For example, gastrin may appear in five different forms in the circulation, due to different splice variants in NETs [54]. Non-functioning NETs are not associated with specific hormonal changes or clinical syndromes. As a result, non-functioning NETs are usually diagnosed in the later stages after the occurrence of symptoms related to tumour mass effects or metastases [50][51]. The liver is the most common site of NET metastasis. Due to improved diagnostic tools and an increase in early diagnosis, the majority of cases at the time of diagnosis are graded as G1; most of these are non-metastatic, but only by a small margin [55].
Biochemical and tissue markers in GEP-NETs are applied for diagnostic, prognostic, and predictive intentions [52][56]. Chromogranin A (CgA) is considered to be one of the most implemented biomarkers in the diagnosis and prognosis of NETs. Despite an overall diagnostic sensitivity of 73% and a specificity of 95%, the use of CgA as a biomarker for NETs has gradually declined in recent years. The accuracy of CgA can vary largely based on the type of NET and it can be falsely elevated in the presence of various conditions, such as atrophic gastritis and liver disease, or in cases involving treatment with proton pump inhibitors [57][58][59][60][61]. Other general markers, including the Neuron-Specific Enolase (NSE) and pancreatic polypeptide (PP), are mainly elevated in poorly differentiated NETs and non-functioning NETs, respectively [56]. The serotonin metabolite 5-hydroxy indole acetic acid (5HIAA) can be measured in urine or blood plasma and is used as a diagnostic and follow-up marker for patients with midgut NETs. However, the specificity of 5HIAA is influenced by the fact that its hypersecretion has also been observed in patients with carcinoid syndrome [62][63][64]. Circulating biomarkers, such as gastrin, insulin, glucagon, SST, and VIP, are specific PanNET biochemical markers used for diagnosis and treatment monitoring [65]. Several novel biomarkers have been discovered to improve the early diagnosis and monitoring of NETs, including programmed death ligand-1 and glucose transporters type 1 within lung NETs (Lu-NETs) and PanNETs, as well as survivin, an inhibitor of apoptosis, in Lu-NETS, PanNETs, and GI-NETs [66]. Circulating tumour cells (CTCs) are a relatively novel biomarker for NETs; the elevation of CTCs is measured based on the expression of the epithelial cell adhesion marker [67][68]. The absence of CTCs strongly correlates with 5HIAA and liver metastases extension, indicating disease progression. Therefore, the absence of CTCs may be considered as a prognostic biomarker [68]. Despite promising results of existing research, additional data and evidence in this regard remain sparse. Further studies are necessary to convincingly demonstrate the clinical usefulness of CTCs as a biomarker for NETs.
Although NETs are mostly sporadic, they can be associated with multiple inherited syndromes, including multiple endocrine neoplasia (MEN) types 1, 2, and 4, as well as Von Hippel–Lindau syndrome, neurofibromatosis 1 (NF1), and tuberous sclerosis. These syndromes are caused by dysregulating mutations in oncogenes, proto-oncogenes, tumour suppressors, or cell cycle regulator genes. As a result, these dysregulations can cause cell tumourous growth, among other symptoms [54][69].
NETs are extensively vascularised tumours. They typically have an increased expression of vascular endothelial growth factor (VEGF) and VEGF receptor (VEGFR) subtypes [46]. Interestingly, VEGF expression has been observed to be higher in well-differentiated NETs, compared to poorer differentiated counterparts. The intratumoural vessel density in NETs is approximately 10-fold higher as compared with carcinomas, which elevates not only NET growth, but also the release of secretory products into the bloodstream [70].
4. Somatostatin Signalling in NET Development and Prognostics
Somatostatin signalling is involved in many cell regulatory circuits that can inhibit tumourigenesis. Therefore, shifts in SST signal transduction pathways can significantly contribute to NET development within an affected tissue. The binding of SST to SSTR inhibits the activity of adenylyl cyclase, downregulating the concentration of the second messenger cAMP and following intracellular calcium, leading to a decreased hormone secretion [71][72]. This is particularly important in hormone-producing NETs, where SSA treatment can help to normalise adverse effects of excessive hormone levels in the body. SST signals are also important cell cycle regulators; a ligand binding to SSTR activates protein tyrosine phosphatases SHP-1 and SHP-2, leading to a decreased cell proliferation via the upregulation of cell cycle inhibitors p27 and p21, as well as the inhibition of PI3K/AKT and MAPK, thereby attenuating cell division [16][71]. This property could indicate the possibility to regulate other tumour growth through targeting the SST signalling pathway, especially since SSTRs are expressed also in breast, thyroid, prostate cancer tissues, glioma, hepatocellular carcinoma, and other tumours [73]. Other hallmarks of tumourigenesis, such as angiogenesis and cell migration, are also regulated by SST signalling. The VEGF platelet-derived growth factor (PDGF), insulin-like growth factor, and basic fibroblast growth factor have been shown to enhance the neovascularization and cell growth of tumours [74][75][76][77]. On a cell signalling and functional level, SST and SSTR significantly inhibit hormonal secretion, cell cycle progression, angiogenesis, and cell migration. However, the role of SST signalling and SSTR in NET development, the response to SSA, and prognosis highly depends upon SSTR distribution in different tumour types and additional intrinsic factors of specific tumours [40].
Many studies have assessed the expression of SSTRs in different tumours. The techniques used in these studies include PCR-based methods, immunohistochemistry (IHC), and somatostatin receptor scintigraphy (SRS), each of which has specific advantages and drawbacks (Table 1) [78][79][80][81][82][83][84][85][86]. PCR-based expression evaluation methods remain cost effective, easy to perform, and are scalable. This method is also easily interpreted without highly sophisticated professional experience or background, compared to IHC and imaging. PCR gives bulk representative values for tumour tissue expression levels. However, in several studies, good concordance has been demonstrated between RT-PCR and IHC data [80][87]. On the other hand, IHC and imaging allow for a more precise evaluation of expression levels. However, these techniques are more costly and are highly dependent on available equipment and professional expertise. In IHC studies, the antibodies used for the evaluation can affect the obtained results [88][89]. Nonetheless, previous reports have successfully reported on the expression of SSTR1, SSTR2A, SSTR3, SSTR4, and SSTR5. In studies where all SSTRs have been assessed simultaneously, it has been demonstrated that SSTR4 is not expressed or is expressed in lower levels in NETs compared to other SSTRs [80][81][87]. The most expressed receptor subtypes are SSTR2A and SSTR5, following by a slightly lower abundance of SSTR3 and SSTR1 [80][81][87][90]. However, this information is still highly dependent upon the tumour type and methods used for the estimation of the expression level. Aside from the methodological differences described above, the subgrouping or classification of tumour types and preoperative SSA treatment can have a significant impact on the obtained results, which can hamper the generalisation of these findings in overarching conclusion.
In many studies, all NET types are analysed together, which is understandable given the rarity of these tumours. However, this can bias the results, since tumour development in specific tissue types causes intrinsic functional differences in tumour cells originating from specific cell types. Additionally, in many reports, the preoperative status of SSA is different; some studies include only SSA-treated patients, whereas others include both SSA-treated and naive cases without properly adjusting for the potential therapeutic impact. It has been shown that SSA has the ability to downregulate SSTR expression, which means that an SSA pre-treatment can significantly affect SSTR expression [83][84].
Nonetheless, it has been widely proven that a higher SSTR expression is characteristic of well-differentiated NETs with tumour grades G1 or G2 [42][48][85][90][91][92]. Additionally, several reports have shown that NET patients with a higher tumour SSTR expression have improved survival [42][78][80][85][86][93][94]. Intriguingly, the better prognosis is also observed in those studies where subjects did not receive SSA therapy or only a small fraction of patients were treated with SSA [42][93][94]. This raises the question of how native SSTR expression without SSA therapy might contribute to better outcomes for NET patients. One plausible answer could be that an SSTR, in the absence of exogenous SSA, still receives endogenous somatostatin signals and this slows the tumour progression. It has been demonstrated that pancreatic NET metastases express lower levels of somatostatin, and the knockdown of somatostatin in pancreatic NET cell lines increases metabolic activity, viability, and growth [95].
In addition to providing prognostic insight on NET development, SSTR expression could also serve as a molecular determinant for predicting the SSA response for personalized therapy choices. This approach has been widely discussed for pituitary NETs, where SSTR expression levels are widely correlated with SSA treatment efficacy [96][97][98]. Other intrinsic tumour components such as genetic predisposition and somatic variation, expression pattern alterations and cellular patterns of dense or sparse granulation can be linked to the SSA response [97][99]. Recently, specific miRNA subtypes have also been shown to be dysregulated by SSA treatment or even downregulate SSTR, promoting tumour progression and indicating the presence of other crucial molecular markers in NETs [100].
The heterodimerization of SSTRs with dopamine receptors has also been widely demonstrated and has the potential to significantly affect NET pathophysiology and prognostics [101]. This has led to the development of chimeric somatostatin–dopamine agonists that could more effectively inhibit tumour progression [102]. Although the first generation of these chimeric compounds demonstrated promising results in preclinical studies, results from human studies were disappointing [101][103][104]. Currently, the second generation of chimeric agonists is under investigation and has shown positive effects both in cell lines and in healthy human trials [105][106][107][108]. Research in the field of chimeric somatostatin–dopamine agonists could bring additional improved therapeutic options for NET patients in the future.
Additionally, in recent years, epigenetic regulation in NETs has been implicated as a major tumourigenesis mechanism. For example, it has been demonstrated that the treatment of pancreatic NET cell lines treated with epigenetic modulators can result in the redifferentiation of human primary PanNETs. The upregulation of SSTR expression was observed in these studies, indicating that SSTR expression can be regulated by epigenetic tumour development mechanisms [109][110]. Specifically, valproic acid was used to upregulate SSTR2 expression and provide further benefit to SSTR-targeted therapies [111]. Other epigenetic agonists have been demonstrated to inhibit cell proliferation, reduce the progression and metastasis-forming capacity, induce apoptosis, and promote cytotoxic effects in various NET cell lines [112][113][114][115]. So far, despite promising evidence in preclinical settings, clinical trials have not confirmed the benefits in patient outcomes. A study conducted on eight patients showed that valproic acid has neutral to moderate effects, with an overall good tolerance to the treatment [116]. However, another study of 15 patients with metastatic NETs demonstrated that the histone deacetylase inhibitor depsipeptide was cardiotoxic [117]. In a separate study of 15 patients receiving Panobinostat, no significant benefits were demonstrated [118]. Taken together, these studies demonstrate that a further investigation of epigenetic agents is needed to determine the best strategy for improving NET control.
Additionally, in some studies, combination therapy using SSA with mTOR inhibitors showed some promising results. In future studies, it is crucial to also study signalling interactions of both pathways in NETs [119]. Overall, SSTR and somatostatin signalling is an important molecular factor that can affect the pathophysiology of NETs and should be considered as a target for SSA therapies for NET treatment.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23031447
References
- Günter Klöppel; Neuroendocrine Neoplasms: Dichotomy, Origin and Classifications. Visceral Medicine 2017, 33, 324-330, 10.1159/000481390.
- Jeffrey S. Chang; Li-Tzong Chen; Yan-Shen Shan; Pei-Yi Chu; Chia-Rong Tsai; Hui-Jen Tsai; An updated analysis of the epidemiologic trends of neuroendocrine tumors in Taiwan. Scientific Reports 2021, 11, 1-10, 10.1038/s41598-021-86839-2.
- Emmanuel Ampofo; Lisa Nalbach; Michael D. Menger; Matthias W. Laschke; Regulatory Mechanisms of Somatostatin Expression. International Journal of Molecular Sciences 2020, 21, 4170, 10.3390/ijms21114170.
- Federico Gatto; Federica Barbieri; Marica Arvigo; Stefano Thellung; Jessica Amarù; Manuela Albertelli; Diego Ferone; Tullio Florio; Biological and Biochemical Basis of the Differential Efficacy of First and Second Generation Somatostatin Receptor Ligands in Neuroendocrine Neoplasms. International Journal of Molecular Sciences 2019, 20, 3940, 10.3390/ijms20163940.
- Mariana Gomes-Porras; Jersy Jair Cárdenas; Cristina Álvarez-Escolá; Somatostatin Analogs in Clinical Practice: A Review. International Journal of Molecular Sciences 2020, 21, 1682, 10.3390/ijms21051682.
- Anna Kathrin Stueven; Antonin Kayser; Christoph Wetz; Holger Amthauer; Alexander Wree; Frank Tacke; Bertram Wiedenmann; Christoph Roderburg; Henning Jann; Somatostatin Analogues in the Treatment of Neuroendocrine Tumors: Past, Present and Future.. International Journal of Molecular Sciences 2019, 20, 3049, 10.3390/ijms20123049.
- Patrik Rorsman; Mark O. Huising; The somatostatin-secreting pancreatic δ-cell in health and disease. Nature Reviews Endocrinology 2018, 14, 404-414, 10.1038/s41574-018-0020-6.
- Rosa Maria Paragliola; Alessandro Prete; Giampaolo Papi; Francesco Torino; Andrea Corsello; Alfredo Pontecorvi; Salvatore Maria Corsello; Clinical utility of lanreotide Autogel® in gastroenteropancreatic neuroendocrine tumors. Drug Design, Development and Therapy 2016, ume 10, 3459-3470, 10.2147/dddt.s76732.
- Balrik Kailey; Martijn van de Bunt; Stephen Cheley; Paul R. Johnson; Patrick MacDonald; Anna Gloyn; Patrik Rorsman; Matthias Braun; SSTR2 is the functionally dominant somatostatin receptor in human pancreatic β- and α-cells. American Journal of Physiology-Endocrinology and Metabolism 2012, 303, E1107-E1116, 10.1152/ajpendo.00207.2012.
- Melissa F. Brereton; Elisa Vergari; Quan Zhang; Anne Clark; Alpha-, Delta- and PP-cells. Journal of Histochemistry & Cytochemistry 2015, 63, 575-91, 10.1369/0022155415583535.
- Stephen Clark; Somatostatin 14. xPharm: The Comprehensive Pharmacology Reference 2007, , , 10.1016/b978-008055232-3.62644-3.
- Stephen Clark; Somatostatin 28. xPharm: The Comprehensive Pharmacology Reference 2007, , , 10.1016/b978-008055232-3.62645-5.
- D J Hadjidakis; S A Raptis; A Souvatzoglou; C Karaiskos; E J Diamantopoulos; S D Moulopoulos; Differences between somatostatin-28 and somatostatin-14 with respect to their biological effects in healthy humans and acromegalics.. Clinical physiology and biochemistry 1986, 4, 372–383, .
- Volker Neugebauer; Mariacristina Mazzitelli; Bryce Cragg; Guangchen Ji; Edita Navratilova; Frank Porreca; Amygdala, neuropeptides, and chronic pain-related affective behaviors. Neuropharmacology 2020, 170, 108052, 10.1016/j.neuropharm.2020.108052.
- Mehtap Cakir; Dorota Dworakowska; Ashley Grossman; Somatostatin receptor biology in neuroendocrine and pituitary tumours: part 1 - molecular pathways. Journal of Cellular and Molecular Medicine 2010, 14, 2570-2584, 10.1111/j.1582-4934.2010.01125.x.
- Eva Costanzi; Carolina Simioni; Ilaria Conti; Ilaria Laface; Gabriele Varano; Cinzia Brenna; Luca M. Neri; Two neuroendocrine G protein‐coupled receptor molecules, somatostatin and melatonin: Physiology of signal transduction and therapeutic perspectives. Journal of Cellular Physiology 2020, 236, 2505-2518, 10.1002/jcp.30062.
- Leo J. Hofland; Steven W. J. Lamberts; The Pathophysiological Consequences of Somatostatin Receptor Internalization and Resistance. Endocrine Reviews 2003, 24, 28-47, 10.1210/er.2000-0001.
- M.J. Klomp; S.U. Dalm; M. de Jong; R.A. Feelders; J. Hofland; Epigenetic regulation of somatostatin and somatostatin receptors in neuroendocrine tumors and other types of cancer. Reviews in Endocrine and Metabolic Disorders 2020, 22, 495-510, 10.1007/s11154-020-09607-z.
- T Reisine; H Kong; K Raynor; H Yano; J Takeda; K Yasuda; G I Bell; Splice variant of the somatostatin receptor 2 subtype, somatostatin receptor 2B, couples to adenylyl cyclase.. Molecular Pharmacology 1993, 44, 1016–1020, .
- David E. Elliott; Jie Li; Arthur M. Blum; Ahmed Metwali; Y. C. Patel; Joel V. Weinstock; SSTR2A is the dominant somatostatin receptor subtype expressed by inflammatory cells, is widely expressed and directly regulates T cell IFN-γ release. European Journal of Immunology 1999, 29, 2454-2463, 10.1002/(sici)1521-4141(199908)29:08<2454::aid-immu2454>3.0.co;2-h.
- Małgorzata Kossut; Agnieszka Łukomska; Grzegorz Dobrzański; Monika Liguz-Lecznar; Receptory somatostatynowe w mózgu. Postępy Biochemii 2018, 64, 213-221, 10.18388/pb.2018_133.
- Naomi R. Latorraca; A. J. Venkatakrishnan; Ron O. Dror; GPCR Dynamics: Structures in Motion. Chemical Reviews 2016, 117, 139-155, 10.1021/acs.chemrev.6b00177.
- Yi Zou; Haiping Tan; Yuanfeng Zhao; Yuan Zhou; Lin Cao; Expression and selective activation of somatostatin receptor subtypes induces cell cycle arrest in cancer cells. Oncology Letters 2018, 17, 1723-1731, 10.3892/ol.2018.9773.
- Thomas Günther; Giovanni Tulipano; Pascal Dournaud; Corinne Bousquet; Zsolt Csaba; Hans-Jürgen Kreienkamp; Amelie Lupp; Marta Korbonits; Justo P Castaño; Hans-Jürgen Wester; et al. International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature. Pharmacological Reviews 2018, 70, 763-835, 10.1124/pr.117.015388.
- Tyralee Goo; Yasutada Akiba; Jonathan D. Kaunitz; Mechanisms of Intragastric pH Sensing. Current Gastroenterology Reports 2010, 12, 465-470, 10.1007/s11894-010-0147-7.
- Andreas Stengel; Yvette Taché; Central somatostatin signaling and regulation of food intake. Annals of the New York Academy of Sciences 2019, 1455, 98-104, 10.1111/nyas.14178.
- Ujendra Kumar; Sneha Singh; Role of Somatostatin in the Regulation of Central and Peripheral Factors of Satiety and Obesity. International Journal of Molecular Sciences 2020, 21, 2568, 10.3390/ijms21072568.
- A M Ten Bokum; L J Hofland; P M Van Hagen; Somatostatin and somatostatin receptors in the immune system: a review.. European Cytokine Network 2000, 11, 161-176, .
- Yehuda Chowers; Liora Cahalon; Maor Lahav; Hagai Schor; Ruth Tal; Simon Bar-Meir; Mia Levite; Somatostatin Through Its Specific Receptor Inhibits Spontaneous and TNF-α- and Bacteria-Induced IL-8 and IL-1β Secretion from Intestinal Epithelial Cells. The Journal of Immunology 2000, 165, 2955-2961, 10.4049/jimmunol.165.6.2955.
- Gisbert Weckbecker; Ian Lewis; Rainer Albert; Herbert A. Schmid; Daniel Hoyer; Christian Bruns; Opportunities in somatostatin research: biological, chemical and therapeutic aspects. Nature Reviews Drug Discovery 2003, 2, 999-1017, 10.1038/nrd1255.
- J. Clay Callison; Ronald C. Walker; Pierre P. Massion; Somatostatin Receptors in Lung Cancer: From Function to Molecular Imaging and Therapeutics. Journal of Lung Cancer 2011, 10, 69-76, 10.6058/jlc.2011.10.2.69.
- Modlin, Irvin M; Öberg, Kjell. A century of advances in neuroendocrine tumor biology and treatment: A tribute to Siegried Oberndorfer; Felsenstein CCCP , 2007: Hannover, Germany, 2007; pp. -.
- V. Barresi; C. Alafaci; F. Salpietro; G. Tuccari; Sstr2A immunohistochemical expression in human meningiomas: Is there a correlation with the histological grade, proliferation or microvessel density?. Oncology Reports 1994, 20, 485-492, 10.3892/or_00000032.
- Jerzy Hankus; Romana Tomaszewska; Neuroendocrine neoplasms and somatostatin receptor subtypes expression. Nuclear Medicine Review 2016, 19, 111-117, 10.5603/nmr.2016.0022.
- Noriko Watanabe; Yoko Nakanishi; Noriko Kinukawa; Sumie Ohni; Yukari Obana; Atsuko Nakazawa; Norimichi Nemoto; Expressions of Somatostatin Receptor Subtypes (SSTR-1, 2, 3, 4 and 5) in Neuroblastic Tumors; Special Reference to Clinicopathological Correlations with International Neuroblastoma Pathology Classification and Outcomes. ACTA HISTOCHEMICA ET CYTOCHEMICA 2014, 47, 219-229, 10.1267/ahc.14024.
- Lisa H. De Vries; Lutske Lodewijk; Stefan M. Willems; Koen Dreijerink; Bart de Keizer; Paul J. Van Diest; Abbey Schepers; Han J. Bonenkamp; Ilse A. C. H. Van Engen-Van Grunsven; Schelto Kruijff; et al. SSTR2A expression in medullary thyroid carcinoma is correlated with longer survival.. Endocrine 2018, 62, 639-647, 10.1007/s12020-018-1706-1.
- Qi Yao; Qianqian Liu; Hui Xu; Zhonghua Wu; Liang Zhou; Zhikai Gu; Peipei Gong; Jianhong Shen; Upregulated Expression of SSTR3 is Involved in Neuronal Apoptosis After Intracerebral Hemorrhage in Adult Rats. Cellular and Molecular Neurobiology 2017, 37, 1407-1416, 10.1007/s10571-017-0471-7.
- Joeri Van Op Den Bosch; Pascal Torfs; Benedicte Y. De Winter; Joris G. De Man; Paul A. Pelckmans; Eric Van Marck; David Grundy; Luc Van Nassauw; Jean-Pierre Timmermans; Effect of genetic SSTR4 ablation on inflammatory peptide and receptor expression in the non-inflamed and inflamed murine intestine. Journal of Cellular and Molecular Medicine 2009, 13, 3283-3295, 10.1111/j.1582-4934.2009.00760.x.
- P Holzer; W Schluet; I T Lippe; W Sametz; Involvement of capsaicin-sensitive sensory neurons in gastrointestinal function.. Acta Physiologica Hungarica 1987, 69, 403-411, .
- Yuheng Hu; Zeng Ye; Fei Wang; Yi Qin; Xiaowu Xu; Xianjun Yu; Shunrong Ji; Role of Somatostatin Receptor in Pancreatic Neuroendocrine Tumor Development, Diagnosis, and Therapy. Frontiers in Endocrinology 2021, 12, 679000, 10.3389/fendo.2021.679000.
- Jacqueline Trouillas; Alexandre Vasiljevic; Marion Lapoirie; Laura Chinezu; Emmanuel Jouanneau; Gérald Raverot; Pathological markers of somatotroph pituitary neuroendocrine tumors predicting the response to medical treatment. Minerva Endocrinologica 2019, 44, 129-136, 10.23736/S0391-1977.18.02933-4.
- Yuhong Wang; Wei Wang; Kaizhou Jin; Cheng Fang; Yuan Lin; Ling Xue; Shiting Feng; Zhiwei Zhou; Chenghao Shao; Minhu Chen; et al. Somatostatin receptor expression indicates improved prognosis in gastroenteropancreatic neuroendocrine neoplasm, and octreotide long-acting release is effective and safe in Chinese patients with advanced gastroenteropancreatic neuroendocrine tumors. Oncology Letters 2017, 13, 1165-1174, 10.3892/ol.2017.5591.
- Adriana G. Ioachimescu; Management of Neuroendocrine Tumors in the Twenty-First Century. Endocrinology and Metabolism Clinics of North America 2018, 47, xiii–xiv, 10.1016/j.ecl.2018.05.005.
- Guido Rindi; David S. Klimstra; Behnoush Abedi-Ardekani; Sylvia L. Asa; Frederik T. Bosman; Elisabeth Brambilla; Klaus J. Busam; Ronald R. De Krijger; Manfred Dietel; Adel K. El-Naggar; et al. A common classification framework for neuroendocrine neoplasms: an International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Modern Pathology 2018, 31, 1770-1786, 10.1038/s41379-018-0110-y.
- Simon Schimmack; Bernhard Svejda; Benjamin Lawrence; Mark Kidd; Irvin M. Modlin; The diversity and commonalities of gastroenteropancreatic neuroendocrine tumors. Langenbeck's Archives of Surgery 2011, 396, 273-298, 10.1007/s00423-011-0739-1.
- Michael S. Lee; Bert H. O’Neil; Summary of emerging personalized medicine in neuroendocrine tumors: are we on track?. Journal of Gastrointestinal Oncology 2016, 7, 804-818, 10.21037/jgo.2016.08.05.
- Frediano Inzani; Gianluigi Petrone; Guido Rindi; The New World Health Organization Classification for Pancreatic Neuroendocrine Neoplasia. Endocrinology and Metabolism Clinics of North America 2018, 47, 463-470, 10.1016/j.ecl.2018.04.008.
- Oana Popa; Sorina Maria Taban; Stelian Pantea; Andrei Dorel Plopeanu; Robert Alexandru Barna; Marioara Cornianu; Anca-Ariana Pascu; Alis Liliana Carmen Dema; The new WHO classification of gastrointestinal neuroendocrine tumors and immunohistochemical expression of somatostatin receptor 2 and 5. Experimental and Therapeutic Medicine 2021, 22, 1-9, 10.3892/etm.2021.10613.
- Azarakhsh Baghdadi; Maryam Ghadimi; Sahar Mirpour; Bita Hazhirkarzar; Mina Motaghi; Timothy M. Pawlik; Ihab R. Kamel; Imaging neuroendocrine tumors: Characterizing the spectrum of radiographic findings. Surgical Oncology 2021, 37, 101529, 10.1016/j.suronc.2021.101529.
- Germano Gaudenzi; Silvia Carra; Alessandra Dicitore; Maria Celeste Cantone; Luca Persani; Giovanni Vitale; Fishing for neuroendocrine tumors. Endocrine-Related Cancer 2020, 27, R163-R176, 10.1530/erc-19-0437.
- M. Beatriz S. Lopes; The 2017 World Health Organization classification of tumors of the pituitary gland: a summary. Acta Neuropathologica 2017, 134, 521-535, 10.1007/s00401-017-1769-8.
- Zu-Yi Ma; Yuan-Feng Gong; Hong-Kai Zhuang; Zi-Xuan Zhou; Shan-Zhou Huang; Yi-Ping Zou; Bowen Huang; Zhong-Hai Sun; Chuan-Zhao Zhang; Yun-Qiang Tang; et al. Pancreatic neuroendocrine tumors: A review of serum biomarkers, staging, and management. World Journal of Gastroenterology 2020, 26, 2305-2322, 10.3748/wjg.v26.i19.2305.
- Irvin M Modlin; Kjell Oberg; Daniel C Chung; Robert T Jensen; Wouter W de Herder; Rajesh Thakker; Martyn Caplin; Gianfranco Delle Fave; Gregory Kaltsas; Eric P Krenning; et al. Gastroenteropancreatic neuroendocrine tumours. The Lancet Oncology 2008, 9, 61-72, 10.1016/s1470-2045(07)70410-2.
- Kjell Öberg; The Genesis of the Neuroendocrine Tumors Concept. Endocrinology and Metabolism Clinics of North America 2018, 47, 711-731, 10.1016/j.ecl.2018.05.003.
- Moises Cukier; Ruth Vergara; Jorge D. Mendez‑Rios; Omar Castillo; Irma Barrera; Eliecer Tello; Olivia El Achtar; Yong Loo; Hector Tapia; Guadalupe Perez; et al. Neuroendocrine tumors in Panama: A nationwide database analysis. Molecular and Clinical Oncology 2021, 15, 1-8, 10.3892/mco.2021.2319.
- George Kanakis; Gregory Kaltsas; Biochemical markers for gastroenteropancreatic neuroendocrine tumours (GEP-NETs). Best Practice & Research Clinical Gastroenterology 2012, 26, 791-802, 10.1016/j.bpg.2012.12.006.
- S. Massironi; M. Fraquelli; S. Paggi; Angelo Sangiovanni; D. Conte; V. Sciola; C. Ciafardini; M. Colombo; M. Peracchi; Chromogranin A levels in chronic liver disease and hepatocellular carcinoma. Digestive and Liver Disease 2009, 41, 31-35, 10.1016/j.dld.2008.05.002.
- Vincenzo Marotta; Maria Chiara Zatelli; Concetta Sciammarella; Maria Rosaria Ambrosio; Marta Bondanelli; Annamaria Colao; Antongiulio Faggiano; Chromogranin A as circulating marker for diagnosis and management of neuroendocrine neoplasms: more flaws than fame. Endocrine-Related Cancer 2018, 25, R11-R29, 10.1530/erc-17-0269.
- Xin Yang; Yuan Yang; Zhilu Li; Chen Cheng; Ting Yang; Cheng Wang; Lin Liu; Shengchun Liu; Diagnostic Value of Circulating Chromogranin A for Neuroendocrine Tumors: A Systematic Review and Meta-Analysis. PLOS ONE 2015, 10, e0124884, 10.1371/journal.pone.0124884.
- Laurent Taupenot; Kimberly L. Harper; Daniel T. O'connor; The Chromogranin–Secretogranin Family. New England Journal of Medicine 2003, 348, 1134-1149, 10.1056/nejmra021405.
- Markos Kalligeros; Leonidas Diamantopoulos; Christos Toumpanakis; Biomarkers in Small Intestine NETs and Carcinoid Heart Disease: A Comprehensive Review. Biology 2021, 10, 950, 10.3390/biology10100950.
- Maria R. Tellez; Gregg Mamikunian; Thomas M. O’Dorisio; Aaron I. Vinik; Eugene A. Woltering; A Single Fasting Plasma 5-HIAA Value Correlates With 24-Hour Urinary 5-HIAA Values and Other Biomarkers in Midgut Neuroendocrine Tumors (NETs). Pancreas 2013, 42, 405-410, 10.1097/mpa.0b013e318271c0d5.
- Louis de Mestier; Frédérique Savagner; Hedia Brixi; Christine Do Cao; Sophie Dominguez-Tinajero; Guillaume Roquin; Bernard Goichot; Olivia Hentic; Olivier Dubreuil; Vincent Hautefeuille; et al. Plasmatic and Urinary 5-Hydroxyindolacetic Acid Measurements in Patients With Midgut Neuroendocrine Tumors: A GTE Study. The Journal of Clinical Endocrinology & Metabolism 2020, 106, 1673-1682, 10.1210/clinem/dgaa924.
- Aart J. Van Der Lely; Wouter W. De Herder; Carcinoid syndrome: diagnosis and medical management. Arquivos Brasileiros de Endocrinologia & Metabologia 2005, 49, 850-860, 10.1590/s0004-27302005000500028.
- Martine Bocchini; Fabio Nicolini; Stefano Severi; Alberto Bongiovanni; Toni Ibrahim; Giorgia Simonetti; Ilaria Grassi; Massimiliano Mazza; Biomarkers for Pancreatic Neuroendocrine Neoplasms (PanNENs) Management—An Updated Review. Frontiers in Oncology 2020, 10, 831, 10.3389/fonc.2020.00831.
- Aura D Herrera-Martínez; Leo J Hofland; María A Gálvez Moreno; Justo P Castaño; Wouter W De Herder; Richard A Feelders; Neuroendocrine neoplasms: current and potential diagnostic, predictive and prognostic markers. Endocrine-Related Cancer 2019, 26, R157-R179, 10.1530/erc-18-0354.
- Kosmas Daskalakis; Olov Norlén; Per Hellman; Peter Stålberg; Applying the use of novel biomarkers for neuroendocrine tumors in the clinic: where are we now?. International Journal of Endocrine Oncology 2019, 6, IJE14, 10.2217/ije-2017-0012.
- Maria Chiara Zatelli; Erika Maria Grossrubatscher; Elia Guadagno; Concetta Sciammarella; Antongiulio Faggiano; Annamaria Colao; Circulating tumor cells and miRNAs as prognostic markers in neuroendocrine neoplasms. Endocrine-Related Cancer 2017, 24, R223-R237, 10.1530/erc-17-0091.
- Mark A. Lewis; Hereditary Syndromes in Neuroendocrine Tumors. Current Treatment Options in Oncology 2020, 21, 1-8, 10.1007/s11864-020-00749-5.
- Mauro Cives; Eleonora Pelle’; Davide Quaresmini; Francesca Maria Rizzo; Marco Tucci; Franco Silvestris; The Tumor Microenvironment in Neuroendocrine Tumors: Biology and Therapeutic Implications. Neuroendocrinology 2019, 109, 83-99, 10.1159/000497355.
- E. Vitali; S. Piccini; G. Trivellin; V. Smiroldo; E. Lavezzi; A. Zerbi; G. Pepe; A.G. Lania; The impact of SST2 trafficking and signaling in the treatment of pancreatic neuroendocrine tumors. Molecular and Cellular Endocrinology 2021, 527, 111226, 10.1016/j.mce.2021.111226.
- Marily Theodoropoulou; Günter K. Stalla; Somatostatin receptors: From signaling to clinical practice. Frontiers in Neuroendocrinology 2013, 34, 228-252, 10.1016/j.yfrne.2013.07.005.
- Hyunjong Lee; Minseok Suh; Hongyoon Choi; Seunggyun Ha; Jin Chul Paeng; Gi Jeong Cheon; Keon Wook Kang; Dong Soo Lee; A pan-cancer analysis of the clinical and genetic portraits of somatostatin receptor expressing tumor as a potential target of peptide receptor imaging and therapy. EJNMMI Research 2020, 10, 1-9, 10.1186/s13550-020-00632-2.
- N. Garcia De La Torre; J. A. H. Wass; H. E. Turner; Antiangiogenic effects of somatostatin analogues. Clinical Endocrinology 2002, 57, 425-441, 10.1046/j.1365-2265.2002.01619.x.
- Uma Rai; Thilini R. Thrimawithana; Celine Valery; Simon Young; Therapeutic uses of somatostatin and its analogues: Current view and potential applications. Pharmacology & Therapeutics 2015, 152, 98-110, 10.1016/j.pharmthera.2015.05.007.
- Marta Annunziata; Raul M. Luque; Mario Durán-Prado; Alessandra Baragli; Cristina Grande; Marco Volante; Manuel D. Gahete; Francesco Deltetto; Marco Camanni; Ezio Ghigo; et al. Somatostatin and somatostatin analogues reduce PDGF-induced endometrial cell proliferation and motility. Human Reproduction 2012, 27, 2117-2129, 10.1093/humrep/des144.
- Sandra Pola; Maria Grazia Cattaneo; Lucia M. Vicentini; Anti-migratory and Anti-invasive Effect of Somatostatin in Human Neuroblastoma Cells. Journal of Biological Chemistry 2003, 278, 40601-40606, 10.1074/jbc.m306510200.
- Philippe Brunner; Ann-Catherine Jörg; Katharina Glatz; Lukas Bubendorf; Piotr Radojewski; Maria Umlauft; Nicolas Marincek; Petar-Marko Spanjol; Thomas Krause; Rebecca A. Dumont; et al. The prognostic and predictive value of sstr2-immunohistochemistry and sstr2-targeted imaging in neuroendocrine tumors. European Journal of Pediatrics 2016, 44, 468-475, 10.1007/s00259-016-3486-2.
- Olivera Casar-Borota; Ansgar Heck; Stefan Schulz; Jahn Marthin Nesland; Jon Ramm-Pettersen; Tove Lekva; Irina Alafuzoff; Jens Bollerslev; Expression of SSTR2a, but not of SSTRs 1, 3, or 5 in Somatotroph Adenomas Assessed by Monoclonal Antibodies Was Reduced by Octreotide and Correlated With the Acute and Long-Term Effects of Octreotide. The Journal of Clinical Endocrinology & Metabolism 2013, 98, E1730-E1739, 10.1210/jc.2013-2145.
- Vito D. Corleto; Massimo Falconi; Francesco Panzuto; Massimo Milione; Ottavia De Luca; Pasquale Perri; Renato Cannizzaro; Cesare Bordi; Paolo Pederzoli; Aldo Scarpa; et al. Somatostatin Receptor Subtypes 2 and 5 Are Associated with Better Survival in Well-Differentiated Endocrine Carcinomas. Neuroendocrinology 2008, 89, 223-230, 10.1159/000167796.
- Evanthia Diakatou; Gregory Kaltsas; Michail Tzivras; George Kanakis; Eugenia Papaliodi; George Kontogeorgos; Somatostatin and Dopamine Receptor Profile of Gastroenteropancreatic Neuroendocrine Tumors: An Immunohistochemical Study. Endocrine Pathology 2011, 22, 24-30, 10.1007/s12022-011-9149-8.
- Evanthia Diakatou; Krystallenia I. Alexandraki; Apostolos V. Tsolakis; George Kontogeorgos; Eleftherios Chatzellis; Anastasia Leonti; Gregory A. Kaltsas; Somatostatin and dopamine receptor expression in neuroendocrine neoplasms: correlation of immunohistochemical findings with somatostatin receptor scintigraphy visual scores. Clinical Endocrinology 2015, 83, 420-428, 10.1111/cen.12775.
- Stine Lyngvi Fougner; Olivera Casar-Borota; Jens P Berg; John K. Hald; Jon Ramm‐Pettersen; Jens Bollerslev; The clinical response to somatostatin analogues in acromegaly correlates to the somatostatin receptor subtype 2a protein expression of the adenoma. Clinical Endocrinology 2007, 68, 458-465, 10.1111/j.1365-2265.2007.03065.x.
- Sanne E. Franck; Federico Gatto; Aart Jan Van Der Lely; Joseph A.M.J.L. Janssen; Alof H.G. Dallenga; A. Paul Nagtegaal; Leo J. Hofland; Sebastian J.C.M.M. Neggers; Somatostatin Receptor Expression in GH-Secreting Pituitary Adenomas Treated with Long-Acting Somatostatin Analogues in Combination with Pegvisomant.. Neuroendocrinology 2016, 105, 44-53, 10.1159/000448429.
- Daniel Kaemmerer; Tina Träger; Maike Hoffmeister; Bence Sipos; Merten Hommann; Jörg Sänger; Stefan Schulz; Amelie Lupp; Inverse expression of somatostatin and CXCR4 chemokine receptors in gastroenteropancreatic neuroendocrine neoplasms of different malignancy. Oncotarget 2015, 6, 27566-27579, 10.18632/oncotarget.4491.
- Shreya Mehta; Philip R. de Reuver; Preetjote Gill; Juliana Andrici; Lisa D’Urso; Anubhav Mittal; Nick Pavlakis; Stephen Clarke; Jaswinder S. Samra; Anthony J. Gill; et al. Somatostatin Receptor SSTR-2a Expression Is a Stronger Predictor for Survival Than Ki-67 in Pancreatic Neuroendocrine Tumors. Medicine 2015, 94, e1281, 10.1097/md.0000000000001281.
- M. Papotti; Massimo Bongiovanni; M. Volante; E. Allìa; Stefania Landolfi; L. Helboe; M. Schindler; S L Cole; G. Bussolati; Expression of somatostatin receptor types 1–5 in 81 cases of gastrointestinal and pancreatic endocrine tumors. Virchows Archiv 2002, 440, 461-475, 10.1007/s00428-002-0609-x.
- Satu M. Remes; Helena L. Leijon; Tiina J. Vesterinen; Johanna T. Arola; Caj H. Haglund; Immunohistochemical Expression of Somatostatin Receptor Subtypes in a Panel of Neuroendocrine Neoplasias. Journal of Histochemistry & Cytochemistry 2019, 67, 735-743, 10.1369/0022155419856900.
- Meike Körner; Véronique Eltschinger; Beatrice Waser; Agnes Schonbrunn; Jean Claude Reubi; Value of Immunohistochemistry for Somatostatin Receptor Subtype sst2A in Cancer Tissues. The American Journal of Surgical Pathology 2005, 29, 1642-1651, 10.1097/01.pas.0000174013.14569.90.
- V. Zamora; A. Cabanne; R. Salanova; C. Bestani; E. Domenichini; F. Marmissolle; N. Giacomi; Juan Manuel Oconnor; G. Méndez; E. Roca; et al. Immunohistochemical expression of somatostatin receptors in digestive endocrine tumours. Digestive and Liver Disease 2010, 42, 220-225, 10.1016/j.dld.2009.07.018.
- L. Righi; M. Volante; V. Tavaglione; A. Billè; L. Daniele; T. Angusti; F. Inzani; G. Pelosi; Guido Rindi; M. Papotti; et al. Somatostatin receptor tissue distribution in lung neuroendocrine tumours: a clinicopathologic and immunohistochemical study of 218 ‘clinically aggressive’ cases. Annals of Oncology 2009, 21, 548-555, 10.1093/annonc/mdp334.
- R. Srirajaskanthan; J. Watkins; L. Marelli; K. Khan; M.E. Caplin; Expression of Somatostatin and Dopamine 2 Receptors in Neuroendocrine Tumours and the Potential Role for New Biotherapies. Neuroendocrinology 2009, 89, 308-314, 10.1159/000179899.
- Kirstine Nielsen; Tina Binderup; Seppo W. Langer; Andreas Kjaer; Pauline Knigge; Veronica Grøndahl; Linea Melchior; Birgitte Federspiel; Ulrich Knigge; P53, Somatostatin receptor 2a and Chromogranin A immunostaining as prognostic markers in high grade gastroenteropancreatic neuroendocrine neoplasms.. BMC Cancer 2020, 20, 27-14, 10.1186/s12885-019-6498-z.
- Kosuke Okuwaki; Mitsuhiro Kida; Tetuo Mikami; Hiroshi Yamauchi; Hiroshi Imaizumi; Shiro Miyazawa; Tomohisa Iwai; Miyoko Takezawa; Makoto Saegusa; Masahiko Watanabe; et al. Clinicopathologic characteristics of pancreatic neuroendocrine tumors and relation of somatostatin receptor type 2A to outcomes. Cancer 2013, 119, 4094-4102, 10.1002/cncr.28341.
- Catherine G. Tran; Aaron T. Scott; Guiying Li; Scott K. Sherman; Po Hien Ear; James R. Howe; Metastatic pancreatic neuroendocrine tumors have decreased somatostatin expression and increased Akt signaling. Surgery 2020, 169, 155-161, 10.1016/j.surg.2020.04.034.
- Eva Venegas-Moreno; Mari C. Vazquez-Borrego; Elena Dios; Noelia Gros-Herguido; Alvaro Flores-Martinez; Esther Rivero-Cortés; Ainara Madrazo-Atutxa; Miguel A. Japón; Raúl M. Luque; Justo P. Castaño; et al. Association between dopamine and somatostatin receptor expression and pharmacological response to somatostatin analogues in acromegaly. Journal of Cellular and Molecular Medicine 2017, 22, 1640-1649, 10.1111/jcmm.13440.
- Antonio C Fuentes-Fayos; Araceli García-Martínez; Aura D Herrera-Martínez; Juan M Jiménez-Vacas; Mari C Vazquez-Borrego; Justo P Castaño; Antonio Picó; Manuel D Gahete; Raúl M Luque; Molecular determinants of the response to medical treatment of growth hormone secreting pituitary neuroendocrine tumors. Minerva Endocrinologica 2019, 44, 109-128, 10.23736/S0391-1977.19.02970-5.
- Annamaria Colao; Renata S. Auriemma; Rosario Pivonello; The effects of somatostatin analogue therapy on pituitary tumor volume in patients with acromegaly. Pituitary 2015, 19, 210-221, 10.1007/s11102-015-0677-y.
- Naoto Yonezawa; Eisuke Nishida; Hikoichi Sakai; Shigeo Koyasu; Fumio Matsuzaki; Kazuko Iida; Ichiro Yahara; Purification and characterization of the 90-kDa heat-shock protein from mammalian tissues. JBIC Journal of Biological Inorganic Chemistry 2005, 177, 1-7, 10.1111/j.1432-1033.1988.tb14337.x-i2.
- Ana Maria Curt; Ioana Rada Popa Ilie; Calin Cainap; Ovidiu Balacescu; Cristina Ghervan; MicroRNAs and Treatment with Somatostatin Analogs in Gastro- Entero-Pancreatic Neuroendocrine Neoplasms: Challenges in Personalized Medicine. Journal of Gastrointestinal and Liver Diseases 2020, 29, 647-659, 10.15403/jgld-2866.
- Maria Cantone; Alessandra Dicitore; Giovanni Vitale; Somatostatin-Dopamine Chimeric Molecules in Neuroendocrine Neoplasms. Journal of Clinical Medicine 2021, 10, 501, 10.3390/jcm10030501.
- Diego Ferone; Alexandru Saveanu; Michael D. Culler; Marica Arvigo; Alberto Rebora; Federico Gatto; Francesco Minuto; Philippe Jaquet; Novel chimeric somatostatin analogs: facts and perspectives. European Journal of Endocrinology 2007, 156, S23-S28, 10.1530/eje.1.02356.
- P Jaquet; G Gunz; A Saveanu; H Dufour; J Taylor; J Dong; S Kim; J-P Moreau; A Enjalbert; M D Culler; et al. Efficacy of chimeric molecules directed towards multiple somatostatin and dopamine receptors on inhibition of GH and prolactin secretion from GH-secreting pituitary adenomas classified as partially responsive to somatostatin analog therapy. European Journal of Endocrinology 2005, 153, 135-141, 10.1530/eje.1.01950.
- Song-Guang Ren; Sun Kim; John Taylor; Josse Dong; Jacques-Pierre Moreau; Michael D. Culler; Shlomo Melmed; Suppression of Rat and Human Growth Hormone and Prolactin Secretion by a Novel Somatostatin/Dopaminergic Chimeric Ligand. The Journal of Clinical Endocrinology & Metabolism 2003, 88, 5414-5421, 10.1210/jc.2003-030302.
- Aura D Herrera-Martínez; Rosanna Van Den Dungen; Fadime Dogan-Oruc; Peter M Van Koetsveld; Michael D. Culler; Wouter W De Herder; Raul M. Luque; Richard A Feelders; Leo J Hofland; Effects of novel somatostatin-dopamine chimeric drugs in 2D and 3D cell culture models of neuroendocrine tumors. Endocrine-Related Cancer 2019, 26, 585-599, 10.1530/erc-19-0086.
- Mari C. Vázquez-Borrego; Fernando L-López; María A. Gálvez-Moreno; Antonio C. Fuentes-Fayos; Eva Venegas-Moreno; Aura D. Herrera-Martínez; Cristóbal Blanco-Acevedo; Juan Solivera; Tanya Landsman; Manuel D. Gahete; et al. A New Generation Somatostatin-Dopamine Analogue Exerts Potent Antitumoral Actions on Pituitary Neuroendocrine Tumor Cells. Neuroendocrinology 2019, 110, 70-82, 10.1159/000500812.
- Alessandra Dicitore; Maria Celeste Cantone; Germano Gaudenzi; Davide Saronni; Silvia Carra; Maria Orietta Borghi; Manuela Albertelli; Diego Ferone; Leo J. Hofland; Luca Persani; et al. Efficacy of a Novel Second-Generation Somatostatin-Dopamine Chimera (TBR-065) in Human Medullary Thyroid Cancer: A Preclinical Study. Neuroendocrinology 2020, 111, 937-950, 10.1159/000512366.
- Wadim M I De Boon; Michiel J Van Esdonk; Frederik E Stuurman; Nienke R Biermasz; Laurent Pons; Isabelle Paty; Jacobus Burggraaf; A Novel Somatostatin-Dopamine Chimera (BIM23B065) Reduced GH Secretion in a First-in-Human Clinical Trial. The Journal of Clinical Endocrinology & Metabolism 2018, 104, 883-891, 10.1210/jc.2018-01364.
- Rosa Schmitz; Julia Weissbach; Jan Kleilein; Jessica Bell; Stefan Hüttelmaier; Fabrice Viol; Till Clauditz; Patricia Grabowski; Helmut Laumen; Jonas Rosendahl; et al. Targeting HDACs in Pancreatic Neuroendocrine Tumor Models. Cells 2021, 10, 1408, 10.3390/cells10061408.
- Mauro Cives; Valeria Simone; Francesca Maria Rizzo; Franco Silvestris; NETs: organ-related epigenetic derangements and potential clinical applications. Oncotarget 2016, 7, 57414-57429, 10.18632/oncotarget.10598.
- Lichun Sun; Qingqing Qian; Guangchun Sun; L. Vienna Mackey; Joseph A. Fuselier; David H. Coy; Cui-Yun Yu; Valproic acid induces NET cell growth arrest and enhances tumor suppression of the receptor-targeted peptide–drug conjugate via activating somatostatin receptor type II. Journal of Drug Targeting 2015, 24, 169-177, 10.3109/1061186x.2015.1066794.
- Yvonne Arvidsson; Viktor Johanson; Roswitha Pfragner; Bo Wängberg; Ola Nilsson; Cytotoxic Effects of Valproic Acid on Neuroendocrine Tumour Cells. Neuroendocrinology 2015, 103, 578-591, 10.1159/000441849.
- Viola Baradari; Alexander Huether; Michael Höpfner; Detlef Schuppan; Hans Scherübl; Antiproliferative and proapoptotic effects of histone deacetylase inhibitors on gastrointestinal neuroendocrine tumor cells. Endocrine-Related Cancer 2006, 13, 1237-1250, 10.1677/erc.1.01249.
- He-Yu Zhang; Kandelaria M. Rumilla; Long Jin; Nobuki Nakamura; Gail A. Stilling; Katharina H. Ruebel; Timothy J. Hobday; Charles Erlichman; Lori A. Erickson; Ricardo V. Lloyd; et al. Association of DNA methylation and epigenetic inactivation of RASSF1A and beta-catenin with metastasis in small bowel carcinoid tumors. Endocrine 2006, 30, 299-306, 10.1007/s12020-006-0008-1.
- Vinita M Alexander; Madhuchhanda Roy; Kristen A Steffens; Muthusamy Kunnimalaiyaan; Herbert Chen; Azacytidine induces cell cycle arrest and suppression of neuroendocrine markers in carcinoids.. International journal of clinical and experimental medicine 2010, 3, 95-102, .
- Tabraiz A. Mohammed; Kyle D. Holen; Renata Jaskula-Sztul; Daniel Mulkerin; Sam J. Lubner; William R. Schelman; Jens Eickhoff; Herbert Chen; Noelle K. LoConte; A Pilot Phase II Study of Valproic Acid for Treatment of Low-Grade Neuroendocrine Carcinoma. The Oncologist 2011, 16, 835-843, 10.1634/theoncologist.2011-0031.
- Manisha H. Shah; Philip Binkley; Kenneth Chan; Jim Xiao; Daria Arbogast; Minden Collamore; Yasser Farra; Donn Young; Michael Grever; Cardiotoxicity of Histone Deacetylase Inhibitor Depsipeptide in Patients with Metastatic Neuroendocrine Tumors. Clinical Cancer Research 2006, 12, 3997-4003, 10.1158/1078-0432.ccr-05-2689.
- Ning Jin; Sam J. Lubner; Daniel L. Mulkerin; Saurabh Rajguru; Lakeesha Carmichael; Herb Chen; Kyle D. Holen; Noelle K. LoConte; A Phase II Trial of a Histone Deacetylase Inhibitor Panobinostat in Patients With Low-Grade Neuroendocrine Tumors. The Oncologist 2016, 21, 785-786g, 10.1634/theoncologist.2016-0060.
- Sara Zanini; Serena Renzi; Francesco Giovinazzo; Giovanna Bermano; mTOR Pathway in Gastroenteropancreatic Neuroendocrine Tumor (GEP-NETs). Frontiers in Endocrinology 2020, 11, 562505, 10.3389/fendo.2020.562505.
- Sara Zanini; Serena Renzi; Francesco Giovinazzo; Giovanna Bermano; mTOR Pathway in Gastroenteropancreatic Neuroendocrine Tumor (GEP-NETs). Frontiers in Endocrinology 2020, 11, 562505, 10.3389/fendo.2020.562505.
This entry is offline, you can click here to edit this entry!