Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
Endothelial cells have a mesodermal origin; during vasculogenesis, a “first draft” of the vascular system is laid down to support the growing embryo. Vascular endothelial cells line the entire circulatory system and show remarkable heterogeneity. Even though endothelial cells originate from the same progenitor cells during development, they eventually contribute to different subtypes of endothelia. Based on morphology, the microvasculature consists of three main phenotypes: discontinuous, fenestrated, and non-fenestrated endothelium.
- endothelial cell
- vascular development
- heterogeneity
1. Introduction
Vascular endothelial cells line the entire circulatory system and show remarkable heterogeneity. Even though endothelial cells originate from the same progenitor cells during development, they eventually contribute to different subtypes of endothelia. Based on morphology, the microvasculature consists of three main phenotypes: discontinuous, fenestrated, and non-fenestrated endothelium (Figure 1). These morphological differences correlate with vascular permeability and contribute to organ-specific functions. Non-fenestrated endothelium has a low permeability, which is found in brain, heart, and lung microvessels, as well as within all larger vessels (i.e., arteries and veins). A fenestrated endothelium has transcellular pores of about 70 nm in diameter [1], which are covered by a thin, non-membranous diaphragm. As fenestrae are associated with increased filtration and trans-endothelial transport functions, they are found in kidney, endocrine glands, and gastric and intestinal mucosa. Lastly, discontinuous endothelium is found exclusively in sinusoidal endothelium, such as the bone marrow, the spleen, and the liver endothelium. The latter has larger fenestrations, up to 100 or 200 nm in diameter, that are devoid of a diaphragm and have large pores within individual cells [1]. Additionally, the underlying basement membrane is only partly developed, resulting in a high permeability. Each organ is made up of different types of endothelia. In the kidney, fenestrated endothelium in the peritubular capillaries and glomeruli ensures proper filtration, while continuous endothelium elsewhere provides the kidney itself with nutrients and oxygen [2]. Similarly, circumventricular organs of the brain are lined with fenestrated endothelium, while elsewhere, the tight blood–brain barrier (BBB) is found [3].
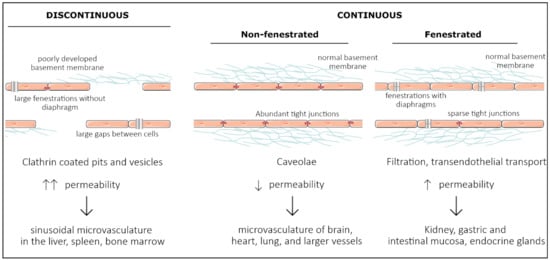
Figure 1. Representation of the three main structural phenotypes in organ-specific microvasculature. Discontinuous endothelium is mainly found in the sinusoidal microvasculature of the liver, spleen and in bone marrow, and is characterized by large fenestrations and pores within and in between endothelial cells, respectively. It has a poorly developed basement membrane and contains clathrin-coated pits and vesicles that dramatically increase permeability. Non-fenestrated endothelium is characterized by low permeability and a high abundance of tight junctions and caveolae. It is mostly found in the microvasculature of the brain, heart, and lung and in larger vessels. Fenestrated endothelium has an intermediate permeability and is characterized by fenestrations covered with a diaphragm. These fenestrations and sparse tight junctions ensure proper filtration and transendothelial transport, as found in the microvasculature of kidney, gastric and intestinal mucosa, and endocrine glands.
2. Vasculogenesis—Intrinsic Versus Extrinsic Factors
Endothelial cells have a mesodermal origin; during vasculogenesis, a “first draft” of the vascular system is laid down to support the growing embryo [10]. In vertebrates, vasculogenesis is initiated in the blood islands at the distal aspect of the yolk sac. The blood islands give rise to both primitive endothelial cells, called angioblasts, and primitive red blood cells, erythroblasts [11,12] (Figure 2). Extraembryonic angioblasts subsequently migrate over the yolk sac to form a randomly organized primitive vascular plexus. VEGF signaling is abundantly studied as a critical mediator of vasculogenesis [13,14,15,16]. Both heterozygous and homozygous Vegf-A null mice died during embryonic development, at E11.5 and E9.5, respectively, due to impaired angiogenesis and disrupted formation of the blood islands [13,14]. Similarly, Vegfr2−/− embryos die between E8.5 and E9.5 due to defects in vasculogenesis and the lack of blood islands [15]. Vasculogenesis is tightly regulated by a family of E26 transformation-specific (ETS) transcription factors [17].
Although a certain redundancy has been described, ETV2 drives both Vegfr2 and Tie2 expression in endothelial progenitor cells [18,19]. Already at E8.5, Etv2−/− embryos present with impaired vasculogenesis and reduced VEGFR2 expression, while they die between E9.0 and E10.5 [18,19]. Although VEGF signaling drives endothelial development, its own expression is regulated by the microenvironment of the developing tissue. In lung tissue, VEGF is initially secreted by mesenchymal cells, while later, the main source shifts to alveolar epithelium [20,21,22]. Between E9.5 and E12, a balance between FGF9 expressed by epithelial cells and FGF10 expressed by the surrounding mesenchymal cells sustains VEGF expression. FGF10 drives mTORC1/Sprouty2 signaling in epithelial cells, which initiates the production of VEGF by the epithelium [23]. In turn, FGF9 binds FGFR1 on epithelial cells and induces the expression of VEGF [24]. Similarly, in the developing heart, cardiomyocytes are an important source of VEGF for the coronary vessels; embryos with a cardiomyocyte-specific Vegf ablation present with fewer coronary vessels and a thinned ventricular wall at E13.5, suggesting that the tissue-specific microenvironment regulates endothelial differentiation by regulating VEGF expression [25]. Furthermore, endothelial differentiation is at least partly driven by FGF2 and BMP4, although they are classically needed to induce the mesoderm. Loss of BMP4, as well as downstream effectors SMAD5 and SMAD4, prevents the induction of mesoderm and as a result, these embryos lack an organized yolk sac vasculature [26,27,28,29]. Moreover, BMP4 alone is sufficient to induce endothelial formation in the absence of endodermal cues, suggesting that BMP4 drives mesodermal formation [30]. However, vasculogenesis alone does not ensure that the vasculature expands, remodels, and adapts according to the requirements of the developing embryo. During development and throughout adult life, new blood vessels are created from the existing vasculature during either sprouting angiogenesis or intussusceptive angiogenesis [31].
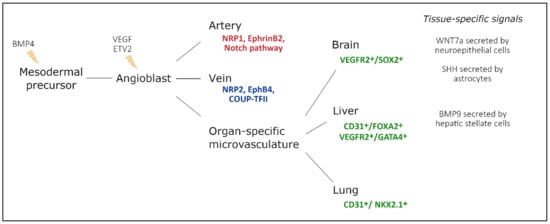
Figure 2. Endothelial cell heterogeneity throughout embryonic development and main factors involved. Mesodermal cells differentiate into vascular and hematopoietic progenitors under the influence of BMP4, secreted by the endoderm. These endothelial progenitor cells differentiate under the influence of VEGF-signaling, driven by ETV2 expression. In turn, they populate arteries, veins, and capillaries. During arteriovenous differentiation, endothelial cells take on an arterial or venous identity, characterized by expression of Nrp1, EphrinB2, and Notch signaling components, or Nrp2, EphB4, and Coup-TFII expression, respectively. Endothelial cells from capillaries are influenced by the microenvironment of their respective organ. Furthermore, a subset of organ-specific endothelial cells also has non-mesodermal origins, based on co-expression of endothelial markers and tissue-specific endoderm markers. Adapted from [32].
3. Large Conduit Vessel Differentiation
Although all endothelial cells originate from the same type of progenitor cell, there is a big discrepancy between large conduit vessels (arteries or veins) and the microvasculature. After the onset of blood flow, the primitive vascular plexus is remodeled into a hierarchical network, including arteries, veins, and capillaries. Although the heart tube already beats by E8.0, proper blood flow is only present once erythrocytes are released from the blood islands at E8.5, with vascular remodeling occurring over the following day [33,34]. In the absence of erythrocytes or blood flow altogether, the primitive plexus fails to remodel into differentiated arteries and veins [33,35]. Moreover, restoring blood viscosity by injecting hetastarch intravascularly in embryos lacking erythrocytes is sufficient to rescue vascular remodeling, highlighting the crucial role of mechanical forces in creating a proper vascular hierarchy [33].
Arterial and venous differentiation is the earliest demonstration of endothelial heterogeneity in the embryo [8]. It is present before the onset of flow [36]; however, shear stress can override this initial identity. Ligation of vitelline arteries in chicken embryos results in a venularization within 24 h, as evidenced by downregulation of Gja5, Nrp1, and EphrinB2 and upregulation of Coup-TFII, Nrp2, and Tie2 [35,37]. Strikingly, reperfusion completely restores the initial arterial gene expression, which highlights the plasticity of endothelial identity and the importance of shear stress for arteriovenous differentiation [35,37].
4. Organ-Specific Vascular Development
As organogenesis commences and tissues specialize into organs with specific morphologies and phenotypes, endothelial cells are no longer exclusively influenced by intrinsic factors. Along embryonic development, newly formed endothelial cells are exposed to extrinsic factors that induce a tissue-specific endothelial differentiation program (Figure 2).
Within the developing brain, WNT7a/b secreted by the neuroepithelial cells binds endothelial WNT receptors Frizzled 4/6/8 and stabilizes β-catenin (encoded by Ctnnb1). Neuroepithelial-specific loss of WNT7a/b signaling induces severe hemorrhages within the central nervous system (CNS) exclusively, demonstrating that the neural microenvironment influences the surrounding vasculature [38]. Endothelial-specific Ctnnb1−/− and Lrp5/6−/− embryos present with a similar phenotype, while the neural tissue completely lacks endothelial cells when endothelial β-catenin is destabilized [39,40]. WNT signaling is thought to drive a specific CNS endothelial differentiation program, including the expression of typical membrane receptor and transporter GLUT1. Impaired WNT7a/b signaling directly reduces GLUT1 expression levels in neural endothelial cells, while ectopic WNT7a drives GLUT1 expression in endothelial cells outside of the CNS [38]. Moreover, in the absence of Ctnnb1, the expression of several typical transcription factors declines in neuronal endothelial cells, demonstrating a key role for β-catenin in activating a BBB-specific differentiation [41].
In contrast to WNT signaling, the effects of GPR124 signaling are confined to specific regions of the developing CNS [42,43]. GPR124 is thought to regulate endothelial cell sprouting and migration in the forebrain and spinal cord exclusively. Gpr124−/− embryos show reduced sprouting in the forebrain region and vascular patterning defects by E11.5 and die by E15.5 [42,43]. GLUT1 expression in the forebrain is reduced in Gpr124−/− embryos, demonstrating that the acquisition of BBB markers may be coupled to developmental brain angiogenesis [42]. Furthermore, sonic hedgehog (SHH) is secreted by perivascular astrocytes and regulates the expression of several tight junction proteins, including Occludin, VE-cadherin, Claudin3, and Claudin5 [44]. Similarly, endothelial-specific loss of Smoothened (Smo), a downstream target of SHH signaling, results in an increased BBB permeability at E14, accompanied by a decreased expression of Occludin, Claudin3, Claudin5, and ZO1 [44].
In the liver, GATA4 has been identified as an important regulator of liver sinusoidal endothelial cell (LSEC) specification [45]. Deleting Gata4 in STAB2+ or LYVE1+ cells results in embryonic lethality between E15.5 and E17.5, while a severely hypoplastic liver is observed at E11.5. LSEC-specific Gata4−/− embryos undergo a major switch from a sinusoidal phenotype to a continuous capillary phenotype, accompanied by the formation of a basement membrane [45]. Furthermore, endothelial VE-Cadherin is upregulated in LSEC-specific Gata4−/− embryos, demonstrating an increased stability of adherens junctions in the liver microvasculature. Recently, BMP9, secreted by hepatic stellate cells that ensheath the liver sinusoids, was identified as one of the paracrine regulators of LSEC fenestration by regulating GATA4 expression in LSECs [46].
Although endothelial cells generally have a mesodermal origin, a subset seems to be derived from tissue-specific endodermal cells. Between E10.5 and E14, a subset of VEGFR2+/SOX2+ endothelial cells is observed within the developing brain, which is lost by E18.5 [47]. Similarly, at E9.5, some rare CD31+FOXA2+ cells are observed in the liver buds; however, by E12.5, they evolve into an evenly dispersed population of CD31+FOXA2+ cells [48]. Furthermore, a subset of GATA4+/VEGFR2+ double-positive cells has been identified between E10.5 and E14 in the budding liver, suggesting a common progenitor cell with hepatocytes [47]. Single-cell analysis of the developing liver predicted that also DLL4, VEGFA, and the TGF-β signaling pathway affect LSEC identity, regulating, amongst others, the expression of Icam2, Sparc, and Lyve1 [49].
Within the lung, a subset of endothelial cells is thought to derive from NKX2.1+ endodermal cells [50]. Already at E10.5, a subset of endothelial cells in the lung co-expresses CD31 and NKX2.1, which is lost by E18.5 [47]. NKX2.1-specific Vegfr2-/- embryos present with a decreased endothelial population [47]. Thus, it seems that a subset of endothelial cells is derived from a similar progenitor type as their surrounding tissue, committing to the vascular lineage over the course of development.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23031477
This entry is offline, you can click here to edit this entry!