Abdominal aortic aneurysm (AAA) is a potentially fatal vascular disease that involves complex multifactorial hemodynamic, thrombotic, inflammatory, and aortic wall remodeling processes. However, its mechanisms are incompletely understood. It has become increasingly clear that platelets are involved in pathological processes of vascular diseases beyond their role in hemostasis and thrombosis. Platelet activation with membrane receptors and secreted mediators promotes thrombus formation and the accumulation of inflammatory cells, which may play an important role in the development of AAA by destroying the structural integrity and stability of the vessel wall. Turbulent blood flow in aortic aneurysms promotes platelet activation and aggregation. Platelet count and heterogeneity are important predictive, diagnostic, and prognostic indicators of AAA.
1. Introduction
Abdominal aortic aneurysm (AAA) is defined as permanent dilatation of the abdominal aorta, which most commonly occurs in the infrarenal region in humans. Although it can have an asymptomatic occurrence, progressive dilation is associated with aortic dissection and rupture [
1]. A population ultrasound screening study reported that the prevalence of AAA is 4–8% in males and 0.5–1.5% in females over the age of 65 [
2]. The current clinical management of AAA focuses on identifying aneurysms while they are asymptomatic and treating them by endovascular aortic aneurysm repair (EVAR) or open surgery. There are no proven pharmaceutical treatments to prevent progressive growth or rupture. Despite improvements in screening and surgical management, the mortality rates of AAA remain high [
3]. A better understanding of AAA development and the emergence of complications is necessary to discover new therapeutic targets.
Abdominal aortic aneurysm is characterized by excessive smooth muscle cell loss, extracellular matrix degradation, and inflammation [
4]. Neutrophils, macrophages, and lymphocytes are the main inflammatory cells in AAA, which secrete collagenase, elastase, and cytokines to promote extracellular matrix degradation and smooth muscle cell apoptosis. The increase in proteolytic activity leads to irreversible remodeling of the aortic wall, resulting in aortic expansion and rupture [
1,
5]. AAA represents a form of atherothrombotic disease, characterized by the formation of a nonocclusive intra-luminal thrombus (ILT) that does not resolve once it occurs. The thromboinflammatory status of ILT contributes to the outward remodeling and eventual disruption of aortic wall integrity [
6,
7].
The major function of platelets is to contribute to hemostasis and thrombosis [
8]. In recent years, many studies have shown that platelets play an important role in the development of AAA [
9]. Low platelet count in patients with AAA suggested an increase in platelet consumption, and ILT formation in aortic aneurysm indicated platelet activation [
10,
11]. Platelet activation participates in AAA pathogenesis via membrane receptors and secreted mediators [
12]. Thrombus formation and the accumulation of inflammatory cells and cytokines in ILT may destroy the structural integrity and stability of the vessel wall, thereby increasing the risk for dissection and rupture [
6]. Disturbances in flow within the aneurysm sac also further promotes platelet activation and aggregation [
13].
2. Targeting Platelet Activation in Abdominal Aortic Aneurysm
2.1. Platelets and Vascular Hemostasis
Platelets are a component of blood whose function is to react to bleeding from blood vessel injury by clumping. Platelets play an important role in the pathophysiology of thrombosis [
14]. Under physiological conditions, thrombus formation on intact endothelial cells is prevented by nitric oxide, adenosine diphosphatase, and prostacyclin. When the endothelial layer is disrupted, collagen and von Willebrand factor (vWF) anchor platelets to the subendothelium. Platelet glycoprotein (GP)Ib/IX/V receptors bind vWF, and GPVI receptors and integrin α2β1 bind collagen. Collagen-mediated GPVI signaling increases the platelet production of thromboxane A2 (TxA2) and decreases the production of prostacyclin [
15]. Activated platelets secrete the contents of granules through their canalicular systems to the exterior, including vWF, platelet factor 4 (PF4), platelet-derived growth factor (PDGF), fibrinogen, coagulation factor V, and β-thromboglobulin from alpha granules, calcium, adenine nucleotides, serotonin, pyrophosphate, and polyphosphate from dense granules, and proteases and glycosidases from lysosomal granules [
16,
17]. Adenosine diphosphate (ADP), vWF, and TxA2 that are released from platelets further promote platelet activation and aggregation [
18]. Clot formation occurs as a result of activating GPIIb/IIIa receptors by changing shape to bind fibrinogen. In addition to this classic mechanism, high-velocity blood flow can also initiate aggregation [
19]. Clinically, platelet activation can be determined by measuring plasma levels of β-thromboglobulin and PF4 [
20].
2.2. Role of Platelets in AAA
2.2.1. Low Platelet Count in AAA
Aortic aneurysm is associated with consumption coagulopathy [
10]. Cases of complicated AAA with chronic disseminated intravascular coagulopathy have been reported [
21,
22]. Chronic disseminated intravascular coagulopathy was cured by surgical repair in an elderly patient with AAA over the next 14 days [
23]. Although no significant differences were found between acutely symptomatic non-ruptured and ruptured AAA [
24], platelet count was significantly lower in patients with AAA compared with healthy controls [
25], suggesting an increase in platelet destruction, most likely through activation within the aneurysm sac [
26,
27]. Patients with AAA had higher baseline spontaneous platelet aggregation compared with normal controls [
28]. The relationship between platelet count and aortic aneurysm size is controversial. A clinical study reported that platelet count decreased as aneurysm size increased, and platelet count was lower in patients with a large AAA (diameter > 55 mm) [
29]. In contrast, no significant differences in platelet count were found between patients with a large AAA and small AAA in another study [
30]. The correlation between platelet indices, such as platelet count, mean platelet volume, the platelet/large cell ratio, and platelet distribution width, are important factors for understanding platelet heterogeneity [
31]. In addition to the decrease in platelet count, mean platelet volume, the mean platelet volume-to-platelet count ratio, the mean platelet volume-to-lymphocyte ratio, and the red cell distribution width-to-platelet count ratio were significantly higher in patients with AAA [
32].
2.2.2. Platelet-Aggregating Thrombus in AAA
Pathophysiological evidence from patients and animal models indicates ILT formation in the lumen in AAA [
33]. The ILT is often structured in three layers in AAA patients: luminal, medial, and abluminal. The luminal ILT layer, which is in contact with blood, is biologically active and enriched in platelets, neutrophils, and red blood cells. The ILT rarely embolizes but does not resolve once it occurs. Eccentric distribution of the ILT was associated with continuous AAA expansion, and a thicker ILT volume was associated with a higher growth rate [
34]. Inflammatory cells and cytokines were reported to accumulate in the ILT and play an important role in AAA progression [
35,
36]. The evolution of ILT can lead to vessel wall weakness through high concentrations of reactive oxygen species, proteases, and cytokines. A study showed that ILT thickness correlated with matrix metalloproteinase 9 (MMP9) expression [
37]. Roxana et al. reported that local C3 retention, consumption, and proteolysis in the ILT could induce polymorphonuclear leukocyte chemotaxis and activation, associated with a decrease in systemic complement concentration and activity in later stages of AAA [
38].
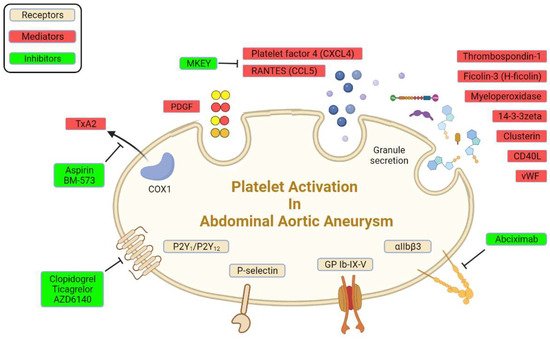
2.3. Platelet Receptors in AAA
There are abundant receptors on the surface of platelets that can bind to the extracellular matrix and adhesion proteins to cause platelet adhesion and activation [
8]. The platelet membrane has several types of receptors, including integrins (αIIbβ3, α2β5, α5β1, and α2β1), leucine-rich receptors (glycoprotein Ib/IX/V and Toll-like receptors), G-protein-coupled receptors (PAR-1, PAR-4, P2Y12, P2Y1, and TxA2), and C-type lectin receptors (P-selectin), among others [
17,
39]. Some of these receptors were reported to interact with various extracellular matrices and cells to accelerate AAA progression (
Figure 1;
Table 1).
Figure 1. Platelet receptors and derived mediators in AAA.
2.3.1. Adenosine Diphosphate Receptors
Adenosine diphosphate is an important activator of platelets [
40]. It exerts its activity through three major purinergic receptors: P2Y12, P2Y1, and P2X1. They are important for changes in platelet shape and aggregation, TxA2 generation, procoagulant activity, adhesion to immobilized fibrinogen, and thrombus formation under shear conditions. P2Y12 receptors are also important for the potentiation of platelet activation that is mediated by other physiological agonists, including collagen, vWF, and TxA2, resulting in sustained platelet activation [
39,
41]. P2Y12 receptor antagonists, such as clopidogrel and ticagrelor, inhibit platelets by selectively and irreversibly binding to P2Y12 receptors and blocking the ADP-dependent pathway of platelet activation. In a rat model that implanted a segment of the sodium dodecyl sulfate-decellularized guinea pig aorta, the P2Y12 receptor antagonist AZD6140 inhibited platelet activation and prevented the development of AAA by inhibiting ADP-induced platelet aggregation and limiting biological activity of the ILT [
42]. Administration of the P2Y12 inhibitor clopidogrel significantly suppressed aortic expansion, elastic lamina degradation, inflammatory cytokine expression, and aortic aneurysm rupture in an established animal model of AAA that was induced by Ang II infusion in hypercholesterolemic mice [
43]. Clopidogrel bisulfate reduced death among patients with AAA [
44]. However, a multicenter randomized, double-blind, controlled trial of ticagrelor and placebo reported different results. Patients who were randomized to ticagrelor did not exhibit a reduction in AAA growth compared with controls during the 12-month follow-up period. This was the first interventional trial on AAA growth using AAA volume as the primary outcome measure other than rupture [
45]. In response to the limited clinical studies of the effects of ADP receptor antagonists on AAA, a Phase 2 clinical trial is currently verifying the efficacy of ticagrelor in patients with a small AAA (ClinicalTrials identifier: NCT02070653).
2.3.2. P-Selectin
P-selectin (CD62P) is an adhesion receptor for neutrophils and macrophages that is expressed on both endothelial cells and platelets [
46]. In platelets, P-selectin is stored in α granules and mobilized to the external plasma membrane within minutes after activation. The expression of P-selectin on activated platelets is important for the recruitment of leukocytes to thrombi and induction of fibrin production during hemostasis. Because detection is relatively easy, soluble P-selectin was assayed as a marker of platelet activity. Soluble P-selectin significantly increased in plasma in patients with AAA [
36]. Likewise, P-selectin significantly increased in an animal model of AAA that was established by xenografting a segment of the sodium dodecyl sulfate-decellularized guinea-pig aorta (xenogenic matrix) onto the abdominal aorta in rats [
36]. P-selectin deficiency attenuated AAA formation in elastase aortic perfusion mice, with diminished aortic wall degradation and preserved elastin and collagen [
47]. P-selectin glycoprotein ligand-1 (PSGL-1) acts as a critical regulator of inflammatory cells infiltration by mediating the adhesion of leukocytes. PSGL-1 deficiency reduced the incidence and severity of AAA by inhibiting inflammatory cell migration and recruitment under conditions of aortic aneurysm [
48].
2.3.3. Other Receptors
Integrin αIIbβ3 is expressed at high levels in platelets and their progenitors. In resting platelets, integrin αIIbβ3 adopts an inactive conformation. Upon agonist stimulation, it switches from a low- to high-affinity state for fibrinogen and other ligands. Ligand binding causes integrin clustering and subsequently promotes outside-in signaling, which drives essential platelet functions, such as spreading, aggregation, clot retraction, and thrombus consolidation [
49]. The inhibition of integrin αIIbβ3 by treatment with the Fab fragment abciximab for 6 weeks reduced both thrombus area and aneurysmal enlargement in a rat xenograft model of AAA compared with treatment with irrelevant immunoglobulins [
36].
GPIb is a major glycoprotein on the platelet surface. Like the integrin αIIbβ3, GPIb undergoes reversible translocation as a function of platelet activation [
50]. As an extramembranous portion of GPIb, glycocalicin was higher in patients with AAA than in patients who underwent carotid endarterectomy, indicating that GPIb was cleaved from the platelet membrane after platelet activation and turnover [
26].
Table 1. Characteristics of studies of platelet receptors in AAA.
| Target |
Inhibitor |
Disease Model |
Study Type |
Main Findings |
Reference |
| ADP receptor |
P2Y12 receptor antagonist AZD6140 |
Decellularized aortic xenograft model of AAA in rats |
Animal study |
Reduced the spontaneous increase in aortic diameter |
[42] |
| ADP receptor |
Clopidogrel |
Apolipoprotein E-knockout mice infused with Ang II (AAA model) |
Animal study |
Suppressed aneurysm formation |
[43] |
| ADP receptor |
Clopidogrel bisulfate |
Hypercholesterolemic mice infused with Ang II (AAA model) |
Animal study |
Reduced AAA rupture |
[44] |
| ADP receptor |
Clopidogrel bisulfate, ticagrelor, or prasugrel |
Patients with AAA who progressed to rupture or dissection |
Cohort study |
Reduced rupture and dissection |
[44] |
| ADP receptor |
Ticagrelor |
Patients with AAA and a maximum aorta diameter of 35–49 mm |
Multicenter randomized controlled trial |
No reduction in growth of small AAA |
[45] |
| P-selectin |
— |
Patients with AAA before surgery |
Cohort study |
Soluble P-selectin significantly increased in plasma |
[36] |
| P-selectin |
— |
Decellularized aortic xenograft model of AAA in rats |
Animal study |
Soluble P-selectin significantly increased in rats |
[36] |
| P-selectin |
Global knockout |
P-selectin knockout mice with elastase perfusion (AAA model) |
Animal study |
P-selectin deficiency attenuated aneurysm formation |
[47] |
| P-selectin |
Global PSGL-1 knockout |
Aortic aneurysm model induced by deoxycorticosterone acetate plus high salt |
Animal study |
Reduced the incidence and severity of aortic aneurysm |
[48] |
| αIIbβ3 |
αIIbβ3 inhibitor abciximab |
Decellularized aortic xenograft model of AAA in rats |
Animal study |
Reduced thrombus area and aneurysmal enlargement |
[36] |
| GPIb |
— |
Patients with asymptomatic AAA |
Case-control study |
Higher glycocalicin produced by cleaved GPIb than normal population |
[26] |
2.5. Platelet Activation and Hemodynamic Changes in AAA
From an engineering perspective, the generation of aortic aneurysm is a failure of the aorta to withstand hemodynamic forces [
76]. Using patient-specific geometries that were derived from computed tomography, computational fluid dynamics has emerged as a powerful and popular tool for studying blood flow dynamics of AAA [
77,
78]. By modeling platelets as infinitesimal and finite-sized particles or even as a continuum quantity, the biomechanical and biochemical activation potential of tracked platelets was quantified.
Much attention has focused on studying hemodynamics in AAA. Using a numerical simulation of flow through an axisymmetric aneurysm under laminar and turbulent steady flow conditions, the recirculation zone formed inside the aneurysm cavity creates conditions that promote platelet deposition and thrombus formation in vitro [
79]. A novel computational particle-hemodynamics analysis of platelet residence times showed high potential to entrap activated blood particles in a patient-specific AAA [
80]. In contrast to the normal aorta, the flow in an aneurysm was highly disturbed. Flow separation that involved regions of high streaming velocities and high shear stress was observed where platelets exhibited adhesion and activation [
19]. Biasetti et al. reported a fluid-dynamics-motivated mechanism of platelet activation, convection, and deposition in AAAs [
81]. A reliable three-dimensional flow visualization method indicated that a longer residence time of recirculated blood flow in the aortic lumen that is caused by this vortex caused sufficient shear-induced platelet activation to develop ILT and maintain uniform flow conditions [
82]. Patient-specific computational fluid dynamic models were used to analyze stress-induced platelet activation within AAA under physiological conditions [
83].
2.6. Clinical Applications Related to Platelets in AAA
2.6.1. Labeled Platelets and Visualization Methods
Labeled platelets and visualization method reveal the role of the platelet activation in aneurysm progression in another way. Accompanied by platelet activation, phosphatidylserine that is exposed on platelet membranes is a mediator that links platelet vesicles to aneurysm progression. Radiolabeled 99mTc-annexin-V specifically binds phosphatidylserine and has been used to assess the renewal activity of ILT in an in vivo experimental model of AAA and ex vivo in human ILT [
84]. 99mTc-fucoidan is an imaging agent for the in vivo detection of biological activity that is associated with P-selectin overexpression on activated platelets in humans and rats with AAA [
85,
86]. Biodegradable microcapsules that are made of polycyanoacrylate and polysaccharide that are functionalized with fucoidan had high binding activities by targeting arterial thrombi that overexpressed P-selectin in human activated platelets and rat AAA thrombotic wall [
87].
2.6.2. Platelets and Surgical Interventions
Surgical repair, including traditional open surgical repair and EVAR, is indicated for AAA with a diameter greater than 5.5 cm in men and 5.0 cm in women, growth of more than 0.5 cm in 6 months, or AAA-related symptoms, such as rupture, dissection, and pain [
88]. Low platelet count at the time of hospital admission predicts poor outcome in patients who undergo the emergency repair of a ruptured AAA [
89,
90]. Platelet count and platelet activity significantly increased after AAA repair [
25,
91]. Platelet count decreased significantly in patients who underwent EVAR during the first few days postsurgery, returning to preoperative levels by 1-week to 1-month post-EVAR [
92,
93,
94]. Vascular surgeons encounter an endovascular-specific problem, the so-called endoleak, which reduces the curability of EVAR. In EVAR in 249 patients, platelet count after EVAR in patients with malignant type II endoleak was lower than in patients without malignant endoleak [
95]. A lack of aneurysm shrinkage by 7 days and 6 months after EVAR was significantly associated with ongoing multiagent antiplatelet therapy with clopidogrel, ticlopidine, cilostazol, and aspirin [
96].
2.6.3. Platelet Infusion and Perioperative Period
Transfusion during open surgery is essential to increase platelet count and function in response to massive blood loss and platelet disorders. Patients with ruptured AAA who received proactive transfusion therapy with platelets had a higher platelet count when they were admitted to the intensive care unit compared with the control group [
97]. Patients with ruptured AAA who received more platelets and plasma intraoperatively had lower 30-day mortality compared with control patients [
98]. Patients who were scheduled to undergo the open repair of a ruptured AAA, however, received no significant benefit from the early administration of platelets with regard to postoperative complications and mortality [
99]. Platelet transfusion was an independent marker of thrombotic complications in patients with ruptured AAA [
100].
This entry is adapted from the peer-reviewed paper 10.3390/biom12020206