Non-Hodgkin lymphoma (NHL) is the most frequent hematological neoplasm in the world with more than 544,000 new NHL cases diagnosed in 2020 (2.8% of all cancer diagnoses). Of all the NHL subtypes, the most common is diffuse large B-cell lymphoma (DLBCL), accounting for approximately 40% of lymphoma cases. DLBCL is also one of the most aggressive subtypes; 5-year survival in elderly patients does not exceed 40%. The most common first-line treatment for DLBCL is chemoimmunotherapy containing rituximab, the so-called R-CHOP regimen (rituximab, cyclophosphamide, doxorubicin, vincristine), which fails in 30–40% of patients. Although relapsed/refractory (r/r) patients receive second-line treatment or may undergo autologous stem cell transplantation, their prognosis remains poor. Years of employment of rituximab as a core of first-line treatment and recent observations on CAR-T cell therapy clearly demonstrate that the use of immunotherapy invariably leads to the induction of resistance.
1. Diffuse Large B-Cell Lymphoma
Non-Hodgkin lymphoma (NHL) is the most frequent hematological neoplasm in the world [
1] with more than 544,000 new NHL cases diagnosed in 2020 (2.8% of all cancer diagnoses) [
2]. Of all the NHL subtypes, the most common is diffuse large B-cell lymphoma (DLBCL), accounting for approximately 40% of lymphoma cases. DLBCL is also one of the most aggressive subtypes; 5-year survival in elderly patients does not exceed 40% [
3].
The most common first-line treatment for DLBCL is chemoimmunotherapy containing rituximab, the so-called R-CHOP regimen (rituximab, cyclophosphamide, doxorubicin, vincristine), which fails in 30–40% of patients. Although relapsed/refractory (r/r) patients receive second-line treatment or may undergo autologous stem cell transplantation, their prognosis remains poor [
4]. As demonstrated by the SCHOLAR-1 trial, the objective response rate (ORR) of r/r patients treated with next-line therapy was 26%, and the median overall survival was 6.3 months [
5]. The greatest challenge in DLBCL therapy is the genetic diversity of this neoplasm, i.e., the presence of mutations, translocations, and chromosomal aberrations. In the face of this heterogeneity, investigation of novel common antigens, which can be targeted by immunotherapy and identified by a universal approach to eliminate malignant cells regardless of their molecular pathogenesis, is constantly pursued [
6].
Recently, r/r patients have gained the access to adoptive therapy using genetically engineered T cells with chimeric antigen receptor (CAR-T cells) targeting DLBCL-expressed surface antigens. Particularly promising results of this treatment strategy have been observed in the case of CD19-CARs. Three of them, i.e., axicabtagene ciloleucel (Yescarta, Kite), lisocabtagene maraleucel (Breyanzi, Bristol Myers Squibb), and tisagenlecleucel (Kymriah, Novartis) are licensed to treat DLBCL patients [
7].
However, years of employment of rituximab as a core of first-line treatment and recent observations on CAR-T cell therapy clearly demonstrate that the use of immunotherapy invariably leads to the induction of resistance [
8]. A deep understanding of the molecular mechanisms underlying this phenomenon is essential for the rational design of novel therapeutic regimens. Therefore, this review aims at summarizing the mechanisms of resistance induced by various forms of immunotherapy used in DLBCL (
Figure 1).
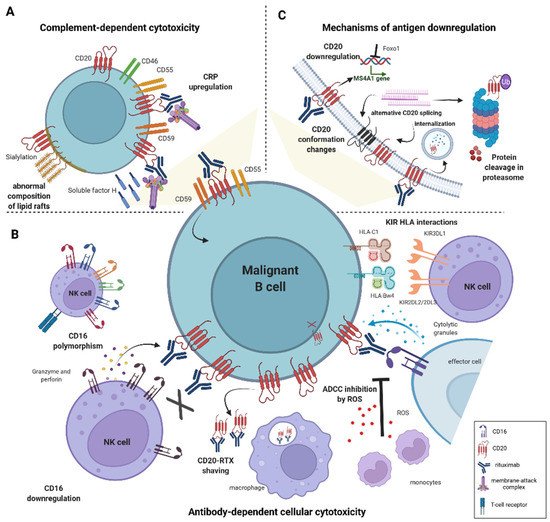
Figure 1. Mechanisms of resistance to anti-CD20-based immunotherapies in DLBCL. (A) Induction of resistance to complement-dependent cytotoxicity (CDC) is mediated by an increased expression of complement regulatory proteins (CRPs)–CD46, CD55, CD59 and soluble protective protein complement factor H (CFH). The abnormal composition of the lipid raft domains, e.g., hypersialylation compromises complement-induced cell lysis. (B) Impaired antibody-dependent cellular cytotoxicity (ADCC) is caused by genetic polymorphism in FcγRIII receptor (CD16) and low expression of CD16. Interactions between specific KIR ligands on NK cells and HLA molecules on B cells reduce NK-cell degranulation. Monocytes mediate shaving of RTX/CD20 complexes from the cell surface; macrophages and neutrophils reduce the anti-tumor potential of NK cells by releasing reactive oxygen species (ROS). (C) CD20 expression is regulated by numerous epigenetic and transcription factors, e.g., Foxo1. Deficiency in CD20 level arises from mutations in MS4A1 gene leading to alternative splicing, and from CD20 internalization. Conformational changes in CD20 protein decrease binding affinity to anti-CD20 mAbs.
2. Rituximab (RTX)
Targeting CD20 antigen using rituximab, a monoclonal antibody (mAb) approved by the FDA in 1997, remains the most widely used form of immunotherapy in oncology. CD20 is a membrane protein found on the surface of normal and neoplastic B lymphocytes, but not on hematopoietic progenitors, which makes it a suitable target for mAbs [
9]. In clinical trials, rituximab combined with chemotherapy significantly improved progression-free survival (PFS) and overall survival (OS) rates in B-cell-derived malignancies such as chronic lymphocytic leukemia (CLL), follicular lymphoma (FL), and DLBCL [
10]. Experience with rituximab provided proof of concept that mAbs could be used in cancer treatment and the combination of rituximab with chemotherapy, i.e., the R-CHOP regimen has remained the first-line of treatment for DLBCL patients for years.
3. Mechanisms of Rituximab Cytotoxicity
The cytotoxic effect of rituximab is exerted through several mechanisms, relying on the activation of anti-tumor immune response (reviewed in [
11,
12]). These include complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and direct apoptosis induction [
13]. Additionally, rituximab sensitizes non-Hodgkin lymphoma cells to chemotherapy [
14,
15]. Rituximab hampers the secretion of interleukin (IL)-10, which is produced by NHL cells as a protective factor. IL-10 induces STAT3 phosphorylation and in consequence the expression of an anti-apoptotic protein Bcl-2, which leads to the resistance to chemotherapeutics [
14,
15]. Moreover, by upregulating Raf-1 kinase inhibitor protein (RKIP), rituximab decreases the signaling of the ERK1/2 and NF-kappa B pathways, leading to downregulation of Bcl-xL [
15]. Furthermore, in NHL cell lines, rituximab inhibits the overexpressed transcription repressor Yin Yang 1 (YY1), which suppresses Fas and DR5 expression, leading to sensitization to Fas ligand and tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced apoptosis [
16].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23031501