Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
The NLRP3 inflammasome is a multiprotein complex that assembles to engage the innate immune defense by processing the maturation of pro-inflammatory cytokines IL-1β and IL-18. Substantial evidence has positioned the NLRP3 inflammasome at the center of vascular disease progression, with a particular significance in the context of aging and the low-grade chronic inflammation associated (inflammaging). Therefore, pharmacological blockade of the NLRP3 inflammasome and its end products has arisen as an extremely promising tool to battle vascular disease.
- NLRP3 inflammasome
- IL-1β
- IL-18
- inflammaging
- pharmacology
1. Introduction
Inflammasomes are a group of cytosolic multiprotein complexes that assemble to engage the innate immune defense by processing the maturation of pro-inflammatory cytokines [1]. Among them, the most studied is the NLRP3 inflammasome. A meaningful role of the NLRP3 inflammasome in age-associated diseases has been evidenced [2], which has paved the way for novel pharmacological interventions to ablate the inflammasome’s effects in neurodegeneration and vascular disease, among others. Multiple mechanisms related to aging and comorbidities such as obesity or diabetes can alter the complex process governing NLRP3 activation, leading to a chronic hyperinflammatory state [3]. In fact, NLRP3 inflammasome over-activation and the subsequent increase in inflammatory cytokines, especially IL-1β, has been frequently linked to atherosclerosis, diabetes, and related chronic sequels [4,5,6].
For all these reasons, understanding the mechanisms regulating NLRP3 inflammasome activation and subsequent signaling is vital to gain insight into potential therapeutic strategies to prevent NLRP3 inflammasome-driven diseases. Furthermore, targeting the NLRP3 inflammasome can offer therapeutic benefits in chronic vascular diseases and subsequent sequels, particularly in light of clinical trials such as CANTOS, which have demonstrated that the inhibition of one of the main products of NLRP3 inflammasome activation, IL-1β, can diminish the risk of major cardiovascular events [7].
2. NLRP3 Inflammasome: Mechanisms and Physiological Function
Priming, assembly and activation of the NLRP3 inflammasome can be triggered by a plethora of pathogenic products and endogenous danger signals sensed by pattern recognition receptors (PRRs) [8]. In a broad context, an array of germline-encoded PRRs mediate the immune surveillance, which can be membrane-bound, such as Toll-like receptors (TLRs) [9]. The NLRP3 inflammasome comprises a sensor protein that belongs to a further set of intracellular PRRs known as the nucleotide-binding oligomerization domain-like receptors (NLRs), including 22 members in humans [8]. NLRP3 receptor is unique among innate immunity elements as it can recognize a vast variety of pathogenic and non-pathogenic endogenous stressors, unlike the majority of PRRs, which have limited specificity to one or few unrelated stimuli [10]. As the sources of NLRP3 inflammasome activation are diverse, this complex could also mediate a heightened state of sterile inflammation when over-activated. Indeed, the NLRP3 inflammasome has been implicated in the pathogenesis of various autoimmune, autoinflammatory, and chronic inflammatory diseases [11].
The NLRP3 inflammasome trimeric complex encompasses several distinct protein/protein interaction domains and acts as a platform to activate the effector protein caspase-1 that prompts the maturation of pro-inflammatory cytokines interleukin (IL)-1β and IL-18, as well as pyroptosis [12]. The complex also includes an adaptor protein with two protein interaction domains, the carboxy-terminal caspase-recruitment domain (CARD) and amino-terminal pyrin domain (PYD), known as the apoptosis-associated speck-like protein containing a CARD (ASC) that bridges the NLRP3 receptor to the procaspase-1 [10]. The NLRP3 protein possesses a PYD domain; a central nucleotide-binding and oligomerization (NACHT) domain that is crucial for the inflammasome assembly and function owing to its ATP-binding properties; and leucine-rich repeats (LRRs), which are thought to be involved in autoinhibition via folding back onto the NACHT domain [10,12,13]. Recent studies also suggest the importance of LRRs and NACHT interaction with the NIMA-related kinase 7 (NEK7) for the activation of the NLRP3 inflammasome [14,15].
Furthermore, the activation of the NLRP3 inflammasome is tightly regulated through a two-step process (Figure 1): (1) priming and (2) assembly and activation [10]. The priming signal initiates a sequence of events to upregulate the insufficient levels of NLRP3 components existing in the cells at the resting state [16]. This transcriptional upregulation can be induced by diverse damaged-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) through various PRRs including TLRs, NLRs, and cytokines’ receptors, which result in nuclear factor-κB (NF-κB) activation and gene transcription of NLRP3 as well as main pro-inflammatory genes, such as pro-IL-1β [16]. During this step, NLRP3 is still controlled through a combination of post-translational modifications (PTMs), which have been identified to be crucial in regulating NLRP3 inflammasome activation [10]. Of these modifications, phosphorylation can induce or inhibit activation depending on the site and stage of NLRP3 activation [17]. Furthermore, ubiquitination and deubiquitination have an essential role in the degradation and activation of NLRP3 [16]. Accordingly, NLRP3 remains in an auto-suppressed inactive state, prepared to receive the activation signal [10,16].
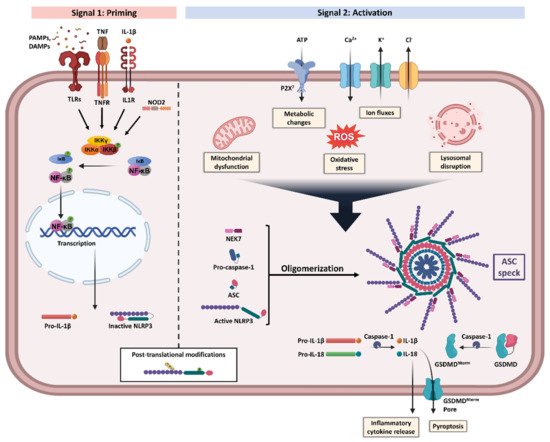
Figure 1. NLRP3 inflammasome priming and activation. NLRP3 inflammasome activation requires two steps, Signal 1 (priming, left) and Signal 2 (assembly and activation, right). Priming is initiated by various exogenous and endogenous stressors that engage pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-containing protein 2 (NOD2), or cytokines receptors like interleukin-1 receptor type 1 (IL-1R1) and tumour necrosis factor receptor (TNFR), leading to nuclear factor-κB (NF-κB) activation and gene transcription of NLRP3 and proIL-1β. Multiple post-translational modifications including ubiquitylation and phosphorylation are crucial for the licensing of the NLRP3 protein activation. Signal 2 is induced by numerous triggers including mitochondrial dysfunction, metabolic changes, oxidative stress, ion fluxes, and lysosomal disruption. The formation of NLRP3 inflammasome protein complex activates caspase-1 that claves pro-IL-1β and pro-IL-18 into their bioactive inflammatory forms. Gasdermin D (GSDMD) is also cleaved and the resulting GSDMD amino terminal (GSDMDNterm) binds to the membrane, forming pores and inducing pyroptosis. Apoptosis-associated speck-like protein containing a CARD (ASC), damaged-associated molecular patterns (DAMPs), IκB kinase (IKK), inhibitor of NF-κB (IκB), NIMA-related kinase 7 (NEK7), P2X purinoceptor 7 (P2X7), pathogen-associated molecular patterns (PAMPs), reactive oxygen species (ROS), and tumour necrosis factor (TNF). Created with BioRender.com. accessed date (15 December 2021).
Multiple cellular signals trigger the activation and assembly of NLRP3 inflammasomes including ion fluxes such as K+ efflux and Ca2+ influx, lysosomal disruption, metabolic changes, mitochondrial dysfunction, and ROS production [6]. However, the roadmap of NLRP3 inflammasome activation is extremely intricated, as many pathways intersect and are interrelated [12]. Once NLRP3 senses the activation signal, it begins the recruitment and oligomerization of ASC and pro-caspase-1 through homophilic interactions to form large speck-like structures [6]. Pro-caspase-1 recruitment enables proximity-induced self-cleavage and activation, producing catalytically active species of caspase-1 [18]. Subsequently, a proteolytic cleavage by caspase-1 generates the bioactive forms of the inflammatory cytokines IL-1β and IL-18 and mediates the activation of the cytosolic protein gasdermin D (GSDMD) [6]. The GSDMD-N domain can bind to membrane lipids and form pores that could mediate the non-conventional secretion of IL-1β and IL-18 and trigger pyroptosis, an inflammatory type of programmed cell death [6,18,19].
3. NLRP3 Inflammasome in Inflammation and Vascular Disease
Inflammation is a major cause underlying vascular diseases. Acute inflammation is triggered by acute events that provoke a systemic response in order to restore homeostasis. However, in the case of chronic inflammation, chronically inflamed tissues produce a prolonged response that induces the migration of leukocytes to the tissues, ultimately producing tissue degeneration and organ damage [20]. With aging, this persistent inflammatory state leads to low-grade sterile chronic inflammation, so-called “inflammaging” [21]. Inflammaging is to date considered a major driver of age-associated diseases and, in particular, of vascular disease.
Both end products of the activation of NLRP3, the cytokines IL-1β and IL-18, are pro-inflammatory and have been associated with several acute and chronic inflammatory diseases, including cardiovascular diseases [22]. IL-1β, among other cytokines and factors, is able to activate the canonical NF-κB signalling pathway [23]. NF-κB is a redox-sensitive transcription factor that, in physiological conditions, is inhibited in the cytoplasm by its binding to IκB. Upon activation, the inhibitor is phosphorylated by the IκB kinase complex and degraded by the proteasome. These events result in the translocation of the subunits p50 and p65 to the nucleus, which activates a variety of genes that are involved in inflammation, among others [24]. In fact, some of the genes regulated by NF-κB are actually pro-IL-1β and pro-IL-18 genes, which code for the precursor proteins cleaved by the inflammasome [25]. Hence, NF-κB is a key modulator of the NLRP3 inflammasome by inducing the priming phase. Besides, IL-1β also induces inflammation by the activation of JNK and p38 [23]. JNK and p38 are mitogen-activated protein kinases (MAPKs), which are phosphorylated and activated by a cascade of MAPK after the binding of IL-1β to its receptor [23]. Once activated, JNK and p38 induce the activation of transcription factors that are involved in the expression of pro-inflammatory genes, such as ATF2 and AP1 [23]. Another consequence of NLRP3 activation is the induction of pyroptosis, a pro-inflammatory cell death leading to an increment in the production and release of IL-1β and IL-18, which contributes to the maintenance of a pro-inflammatory environment [26] and has been demonstrated to contribute to vascular disease [27].
Atherosclerosis is the main cause underlying vascular disease [28] and there are several pieces of evidence pointing out that inflammation and NLRP3 play an important role in this disease [29]. Oxidized LDL (oxLDL), which is known to accumulate in the vessel wall during atherosclerosis and is phagocyted by macrophages, activates NLRP3 by processes related to influx of Ca2+, reactive oxygen species, and mitochondrial dysfunction, among others [27]. Moreover, it is known that cholesterol crystals, which are also present in atherosclerotic lesions, can be phagocyted by human macrophages and induce the production and secretion of IL-1β by the activation of NLRP3 inflammasome, triggering inflammation [30]. There is also a study in which the injection of cholesterol crystals to control mice induced inflammation, while it had no inflammatory effects in mice deficient in NLRP3 components, assessing the important role of NLRP3 components in inflammatory processes in vivo [31].
In two models of atherosclerosis, ApoE−/− mice with high-fat diet and ApoE−/− mice with high-fat and high-methionine diet, the levels of IL-1β and IL-18, macrophage infiltration and atherosclerotic lesions were increased compared with control mice, while the silencing of NLRP3 gene reduced all of these effects [32]. Moreover, NLRP3 inflammasome proteins and its produced cytokines have been shown to be over-expressed in human carotid atherosclerotic plaques, especially in unstable plaques [5]. Over-activated NLRP3 inflammasome was also related to atherosclerosis in diabetic patients, and NLRP3 knockdown reduced and stabilized atherosclerotic plaque in a diabetic atherosclerosis mouse model [33].
IL-1β is known to be produced by hematopoietic cells, as well as vascular endothelial and smooth muscle cells under inflammatory conditions, in which they also induce proliferation and have inflammatory effects contributing to the process of atherosclerosis [5]. Moreover, IL-1β is known to provoke the expression of adhesion molecules in vascular endothelial cells, which results in the migration of monocytes and other leukocytes [34]. Moreover, the other end product of NLRP3, IL-18, also contributes to atherogenesis. The arterial tissue from patients with atherosclerosis showed an increased expression of IL-18 and its receptor compared with healthy tissue [35]. Hence, it has been demonstrated that mononuclear macrophages present in atherosclerotic lesions express IL-18, and endothelial and vascular smooth muscle cells (VSMCs) as well as mononuclear macrophages express the IL-18 receptor [35]. Furthermore, the deficiency of IL-18 in a mice model of atherosclerosis showed decreased atherosclerotic lesions [36]. All of these findings reinforce the significant role of NLRP3 and its derived cytokines in the pathogenesis of atherosclerosis.
The detrimental role of NLRP3 inflammasome and IL-1β from VSMCs in atherosclerosis has recently been challenged by a study from Gomez et al., showing some atheroprotective effects of IL-1β [37]. This study shows smaller lesions from SMC-specific IL1r1−/− Apoe−/− mice, but also reports signs of instability in these animals and SMC-lineage tracing Apoe−/− mice with advanced atherosclerosis treated with anti-IL-1β. A considerable number of studies have reported that NLRP3 inflammasome governs the pro-inflammatory phenotypic switch of VSMCs during atherogenesis [38,39], and contributes to vascular inflammation [40,41], possibly favouring plaque instability and disease progression. Besides, the CANTOS study has provided a great body of evidence on the cardiovascular benefits of anti-IL-1β therapy. Anyhow, this trial also showed inhibition of beneficial outward remodelling on treated patients, and this, together with the work from Gomez et al. [37], highlights the potential adverse effect of excessive IL-1β inhibition within VSMCs. It is important to note that the results by Gomez et al. [37] were obtained under extreme inhibitory conditions, such as the IL1r1 knockout mice, and these might not reflect the reduction circumstances from a human drug treatment.
From a clinical point of view, NLRP3 inflammasome activation has been suggested as a relevant indicator of cardiovascular disease severity and quantification of its components and end products in circulating cells and plasma/serum arise as novel potential biomarkers. Regarding coronary atherosclerosis, NLRP3 is thought to be correlated with the severity of acute coronary syndrome (ACS); that is patients with ACS present higher levels of NLRP3 protein in peripheral blood monocytes and higher levels of IL-1β and IL-18 in plasma than control patients, and the levels of NLRP3 increased with the severity of the disease [42]. Another study demonstrates that the levels of caspase-1 are higher in aortas from patients with coronary disease compared with healthy patients, and that those levels also correlate with the severity of the disease [43]. Moreover, in both studies, it has been observed that patients with typical cardiovascular disease risk factors present higher levels of NLRP3 inflammasome activation [42,43].
NLRP3 has not only been linked to atherosclerosis, but also to other harmful vascular processes, such as abdominal aortic aneurysm (AAA). There is a study in which the expression of NLRP3 was analysed by means of ASC and caspase-1 protein levels from biopsies of patients suffering AAA, reporting increased levels of expression compared with healthy patients [44].
In the context of diabetes, hyperglycemia would lead to mitochondrial dysfunction and oxidative stress, as well as chronic inflammation, all of which shall induce the NLRP3 inflammasome [45]. In fact, in drug-naïve type 2 diabetic patients, an increase in the expression of inflammasome components NLRP3 and ASC was observed in monocyte-derived macrophages, as well as higher serum levels of IL-1β and IL-18 [46]. In line with these results, a diabetic rat model showed excessive activation of NLRP3 inflammasome accompanied by cardiac inflammation, pyroptosis, and fibrosis, which were suppressed by NLRP3 gene silencing therapy [47].
Although IL-1β has been more extensively studied, IL-18 and a cytokine released downstream of both end products of NLRP3 inflammasome, IL-6, both play a crucial role in vascular pathogenesis, and pharmacological interventions targeting these two interleukins are emerging.
This entry is adapted from the peer-reviewed paper 10.3390/antiox11020269
This entry is offline, you can click here to edit this entry!