Auditory Neuropathy (AN) is characterized by disruption of temporal coding of acoustic signals in auditory nerve fibers resulting in alterations of auditory perceptions. Mutations in several genes have been associated to the most forms of AN. Underlying mechanisms include both pre-synaptic and post-synaptic damage involving inner hair cell (IHC) depolarization, neurotransmitter release, spike initiation in auditory nerve terminals, loss of auditory fibers and impaired conduction. In contrast, outer hair cell (OHC) activities (otoacoustic emissions [OAEs] and cochlear microphonic [CM]) are normal. Disordered synchrony of auditory nerve activity has been suggested as the basis of both the alterations of auditory brainstem responses (ABRs) and reduction of speech perception. Authors will review how electrocochleography (ECochG) recordings provide detailed information to help objectively define the sites of auditory neural dysfunction and their effect on receptor summating potential (SP) and neural compound action potential (CAP), the latter reflecting disorders of ribbon synapses and auditory nerve fibers.
- OPA1-related deafness
- OTOF-related hearing loss
- electrocochleography
- cochlear implants
- speech perception
1. Introduction
| Locus | Gene | Transmission | Phenotype | Reference | |
|---|---|---|---|---|---|
| Isolated AN | |||||
| 2p23–p22 | OTOF | Recessive | Congenital profound deafness | [13] | |
| 2q31.1–q31.3 | PJVK | Recessive | Congenital profound deafness | [14] | |
| 13q21–q24 | DIAPH3 | Dominant | Moderate to profound deafness | [15] | |
| mtDNA | 12S rRNA (T1095C) | Moderate deafness | [16] | ||
| 12q23.1 | SLC17A8 | Dominant | Progressive | [17] | |
| 3P25.1 | TMEM43 | Dominant | Post-lingual moderate to profound deafness | [18] | |
| Non-isolated AN | |||||
| CMT 1A | 17p11.2–p12 | PMP22 | Dominant | Mild to severe deafness; demyelinating neuropathy | [19] |
| CMT 1B | 1q22 | MPZ | Dominant | Mild to severe deafness; demyelinating neuropathy | [20] |
| CMT 2E | 8p21 | NF-L | Dominant | Normal hearing; axonal neuropathy | [21] |
| CMT 4D | 8q24.3 | NDRG1 | Recessive | Mild to severe deafness; axonal/demyelinating neuropathy | [6][22] |
| CMT | 1p34 | GJB3 (Cx31) | Dominant | Mild deafness | [23] |
| CMT 1X | Xp13 | GJB1 (Cx32) | X-linked Dominant | Demyelinating neuropathy | [24] |
| ADOA | 3q28–q29 | OPA1 (R445H) | Dominant | Optic neuropathy; moderate deafness | [25] |
| ADOA | 16q21–q22 | Dominant | Optic neuropathy, cardiac abnormalities | [26] | |
| AROA | 11q14.1–11q22.3 | TMEM126A | Recessive | Optic neuropathy; mild hearing loss | [27] |
| Friedreich | 9q13 | FXN | Recessive | Ataxia; axonal neuropathy; optic neuropathy; cardiomyopathy; normal hearing threshold-mild deafness | [28] |
| AUNX1 | Xq23–q27.3 | X-linked Recessive | Sensory axonal neuropathy; mild-to-severe deafness | [29] | |
| DDON (Mohr-Tranebjaerg) | Xq22.1 | TIMM8A | X-linked Recessive | Progressive deafness; dystonia, optic neuropathy; dementia | [30] |
| LHON (Leber) | mtDNA | MTND4 (11778mtDNA) | Optic neuropathy; mild-to-moderate deafness | [31] | |
| Perrault | 10q24.31 | TWNK | Recessive | Hypogonadism, cerebellar atrophy, cochlear nerve thinning | [32] |
| USH3A | 3q25.1 | CLRN1 | Recessive | Retinitis pigmentosa | [33] |
| CAPOS | 19q13.2 | ATP1A3 | Dominant | Cerebellar ataxia, areflexia, pes cavus, optic atrophy | [34] |
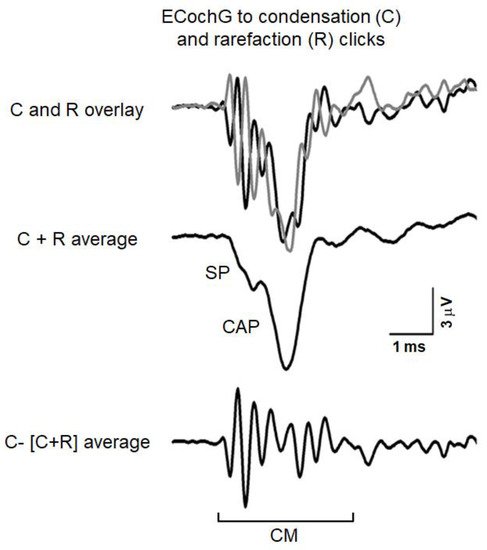
2. Electrocochleography (ECochG)
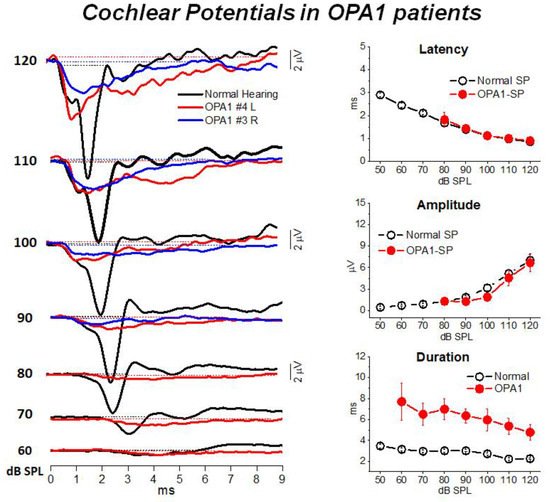
3. Mutations in the OPA1 Gene: A Model of Post-Synaptic AN
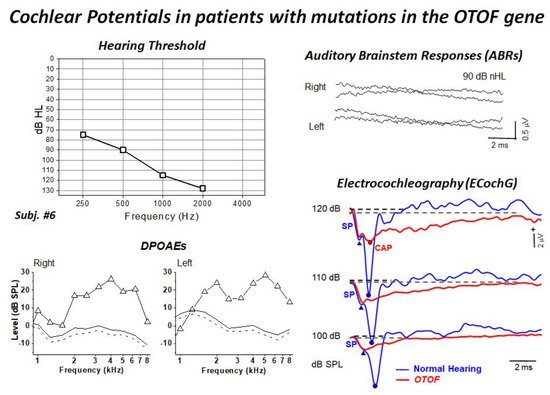
4. Mutations in the OTOF Gene: A Model of Synaptopathy
This entry is adapted from the peer-reviewed paper 10.3390/audiolres11040059
References
- Starr, A.; Picton, T.W.; Sininger, Y.; Hood, L.J.; Berlin, C.I. Auditory neuropathy. Brain 1996, 119, 741–753.
- Starr, A.; Zeng, F.; Michalewski, H.; Moser, T. Perspectives on auditory neuropathy: Disorders of inner hair cell, auditory nerve, and their synapse. In The Senses: A Comprehensive Reference; Basbaum, A.I., Kaneko, A., Shepherd, G.M., Westheimer, G., Albright, T.D., Masland, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; Volume 3: Audition, pp. 397–412.
- Zeng, F.-G.; Kong, Y.-Y.; Michalewski, H.J.; Starr, A. Perceptual Consequences of Disrupted Auditory Nerve Activity. J. Neurophysiol. 2005, 93, 3050–3063.
- Moser, T.; Starr, A. Auditory neuropathy—Neural and synaptic mechanisms. Nat. Rev. Neurol. 2016, 12, 135–149.
- Johnson, J.S.; Newport, E.L. Critical period effects on universal properties of language: The status of subjacency in the acquisition of a second language. Cognition 1991, 39, 215–258.
- Butinar, D.; Zidar, J.; Leonardis, L.; Popovic, M.; Kalaydjieva, L.; Angelicheva, D.; Sininger, Y.; Keats, B.; Starr, A. Hereditary auditory, vestibular, motor, and sensory neuropathy in a Slovenian Roma (Gypsy) kindred. Ann. Neurol. 1999, 46, 36–44.
- Santarelli, R. Information from cochlear potentials and genetic mutations helps localize the lesion site in auditory neuropathy. Genome Med. 2010, 2, 91.
- Santarelli, R.; La Morgia, C.; Valentino, M.L.; Barboni, P.; Monteleone, A.; Scimemi, P.; Carelli, V. Hearing Dysfunction in a Large Family Affected by Dominant Optic Atrophy (OPA8-Related DOA): A Human Model of Hidden Auditory Neuropathy. Front. Neurosci. 2019, 13, 501.
- Santarelli, R.; del Castillo, I.; Cama, E.; Scimemi, P.; Starr, A. Audibility, speech perception and processing of temporal cues in ribbon synaptic disorders due to OTOF mutations. Hear. Res. 2015, 330, 200–212.
- Santarelli, R.; Scimemi, P.; Costantini, M.; Domínguez-Ruiz, M.; Rodríguez-Ballesteros, M.; del Castillo, I. Cochlear Synaptopathy due to Mutations in OTOF Gene May Result in Stable Mild Hearing Loss and Severe Impairment of Speech Perception. Ear Hear. 2021, 42, 1627–1639.
- Huang, T.; Santarelli, R.; Starr, A. Mutation of OPA1 gene causes deafness by affecting function of auditory nerve terminals. Brain Res. 2009, 1300, 97–104.
- Santarelli, R.; Rossi, R.; Scimemi, P.; Cama, E.; Valentino, M.L.; La Morgia, C.; Caporali, L.; Liguori, R.; Magnavita, V.; Monteleone, A.; et al. OPA1-related auditory neuropathy: Site of lesion and outcome of cochlear implantation. Brain 2015, 138, 563–576.
- Rodríguez-Ballesteros, M.; del Castillo, F.J.; Martín, Y.; Moreno-Pelayo, M.A.; Morera, C.; Prieto, F.; Marco, J.; Morant, A.; Gallo-Terán, J.; Morales-Angulo, C.; et al. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF). Hum. Mutat. 2003, 22, 451–456.
- Delmaghani, S.; del Castillo, F.; Michel, V.; Leibovici, M.; Aghaie, A.; Ron, U.; Van Laer, L.; Ben-Tal, N.; Van Camp, G.; Weil, D.; et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat. Genet. 2006, 38, 770–778.
- Schoen, C.J.; Emery, S.B.; Thorne, M.C.; Ammana, H.R.; Sliwerska, E.; Arnett, J.; Hortsch, M.; Hannan, F.; Burmeister, M.; Lesperance, M. Increased activity of Diaphanous homolog 3 (DIAPH3)/diaphanous causes hearing defects in humans with auditory neuropathy and in Drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 13396–13401.
- Wang, Q.J.; Li, Q.Z.; Rao, S.Q.; Lee, K.; Huang, X.S.; Yang, W.Y.; Zhai, S.Q.; Guo, W.W.; Guo, Y.F.; Yu, N.; et al. AUNX1, a novel locus responsible for X linked recessive auditory and peripheral neuropathy, maps to Xq23-27.3. J. Med. Genet. 2005, 43, e33.
- Thirlwall, A.S.; Brown, D.J.; McMillan, P.M.; Barker, S.E.; Lesperance, M.M. Phenotypic Characterization of Hereditary Hearing Impairment Linked to DFNA25. Arch. Otolaryngol.-Head Neck Surg. 2003, 129, 830–835.
- Jang, M.W.; Oh, D.-Y.; Yi, E.; Liu, X.; Ling, J.; Kim, N.; Sharma, K.; Kim, T.Y.; Lee, S.; Kim, A.-R.; et al. A nonsense TMEM43 variant leads to disruption of connexin-linked function and autosomal dominant auditory neuropathy spectrum disorder. Proc. Natl. Acad. Sci. USA 2021, 118, e2019681118.
- Kovach, M.; Campbell, K.; Herman, K.; Waggoner, B.; Gelber, D.; Hughes, L.; Kimonis, V. Anticipation in a unique family with Charcot-Marie-Tooth syndrome and deafness: Delineation of the clinical features and review of the literature. Am. J. Med. Genet. 2002, 108, 295–303.
- Starr, A.; Michalewski, H.J.; Zeng, F.; Fujikawa-Brooks, S.; Linthicum, F.; Kim, C.S.; Winnier, D.; Keats, B. Pathology and physiology of auditory neuropathy with a novel mutation in the MPZ gene (Tyr145->Ser). Brain 2003, 126, 1604–1619.
- Butinar, D.; Starr, A.; Zidar, J.; Koutsou, P.; Christodoulou, K. Auditory nerve is affected in one of two different point mutations of the neurofilament light gene. Clin. Neurophysiol. 2008, 119, 367–375.
- Kalaydjieva, L.; Gresham, D.; Gooding, R.; Heather, L.; Baas, F.; de Jonge, R.; Blechschmidt, K.; Angelicheva, D.; Chandler, D.; Worsley, P.; et al. N-myc Downstream-Regulated Gene 1 Is Mutated in Hereditary Motor and Sensory Neuropathy–Lom. Am. J. Hum. Genet. 2000, 67, 47–58.
- Lopez-Bigas, N.; Olive, M.; Rabionet, R.; Ben-David, O.; Martínez-MatosJ, A.; Bravo, O.; Banchs, I.; Volpini, V.; Gasparini, P.; Avraham, K.B.; et al. Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment. Hum. Mol. Genet. 2001, 10, 947–952.
- Bähr, M.; Andres, F.; Timmerman, V.; E Nelis, M.; Van Broeckhoven, C.; Dichgans, J. Central visual, acoustic, and motor pathway involvement in a Charcot-Marie-Tooth family with an Asn205Ser mutation in the connexin 32 gene. J. Neurol. Neurosurg. Psychiatry 1999, 66, 202–206.
- Amati-Bonneau, P.; Guichet, A.; Olichon, A.; Chevrollier, A.; Viala, F.; Miot, S.; Ayuso, C.; Odent, S.; Arrouet, C.; Verny, C.; et al. OPA1 R445H mutation in optic atrophy associated with sensorineural deafness. Ann. Neurol. 2005, 58, 958–963.
- Carelli, V.; Schimpf, S.; Fuhrmann, N.; Valentino, M.L.; Zanna, C.; Iommarini, L.; Papke, M.; Schaich, S.; Tippmann, S.; Baumann, B.; et al. A clinically complex form of dominant optic atrophy (OPA8) maps on chromosome 16. Hum. Mol. Genet. 2011, 20, 1893–1905.
- Meyer, E.; Michaelides, M.; Tee, L.J.; Robson, A.; Rahman, F.; Pasha, S.; Luxon, L.M.; Moore, A.T.; Maher, E.R. Nonsense mutation in TMEM126A causing autosomal recessive optic atrophy and auditory neuropathy. Mol. Vis. 2010, 16, 650–664.
- Rance, G.; Fava, R.; Baldock, H.; Chong, A.; Barker, E.; Corben, L.; Delatycki, M.B. Speech perception ability in individuals with Friedreich ataxia. Brain 2008, 131, 2002–2012.
- Wang, Q.; Li, R.; Zhao, H.; Peters, J.L.; Liu, Q.; Yang, L.; Han, D.; Greinwald, J.H., Jr.; Young, W.-Y.; Guan, M.-X. Clinical and molecular characterization of a Chinese patient with auditory neuropathy associated with mitochondrial 12S rRNA T1095C mutation. Am. J. Med. Genet. Part A 2005, 133A, 27–30.
- Bahmad, F.; Merchant, S.N.; Nadol, J.B.; Tranebjaerg, L. Otopathology in Mohr-Tranebjaerg syndrome. Laryngoscope 2007, 117, 1202–1208.
- Ceranić, B.; Luxon, L.M. Progressive auditory neuropathy in patients with Leber’s hereditary optic neuropathy. J. Neurol. Neurosurg. Psychiatr. 2004, 75, 626–630.
- Ołdak, M.; Oziębło, D.; Pollak, A.; Stępniak, I.; Lazniewski, M.; Lechowicz, U.; Kochanek, K.; Furmanek, M.; Tacikowska, G.; Plewczynski, D.; et al. Novel neuro-audiological findings and further evidence for TWNK involvement in Perrault syndrome. J. Transl. Med. 2017, 15, 25.
- Dulon, D.; Papal, S.; Patni, P.; Cortese, M.; Vincent, M.; Tertrais, M.; Emptoz, A.; Tlili, A.; Bouleau, Y.; Michel, V.; et al. Clarin-1 gene transfer rescues auditory synaptopathy in model of Usher syndrome. J. Clin. Investig. 2018, 128, 3382–3401.
- Tranebjærg, L.; Strenzke, N.; Lindholm, S.; Rendtorff, N.D.; Poulsen, H.; Khandelia, H.; Kopec, W.; Lyngbye, T.J.B.; Hamel, C.; Delettre, C.; et al. The CAPOS mutation in ATP1A3 alters Na/K-ATPase function and results in auditory neuropathy which has implications for management. Qual. Life Res. 2018, 137, 111–127.
- Santarelli, R.; Starr, A.; Michalewski, H.J.; Arslan, E. Neural and receptor cochlear potentials obtained by transtympanic electrocochleography in auditory neuropathy. Clin. Neurophysiol. 2008, 119, 1028–1041.
- Santarelli, R.; Arslan, E. Electrocochleography. In Handbook of Clinical Neurophysiology; Celesia, G.G., Ed.; Disorders of Peripheral and Central Auditory Processing; Elsevier: Amsterdam, The Netherlands, 2013; pp. 83–113.
- Dallos, P.; Cheatham, M.A. Production of cochlear potentials by inner and outer hair cells. J. Acoust. Soc. Am. 1976, 60, 510–512.
- Durrant, J.D.; Wang, J.; Ding, D.L.; Salvi, R.J. Are inner or outer hair cells the source of summating potentials recorded from the round wi‘ndow? J. Acoust. Soc. Am. 1998, 104, 370–377.
- Pappa, A.K.; Hutson, K.A.; Scott, W.C.; Wilson, J.D.; Fox, K.E.; Masood, M.M.; Giardina, C.K.; Pulver, S.H.; Grana, G.D.; Askew, C.; et al. Hair cell and neural contributions to the cochlear summating potential. J. Neurophysiol. 2019, 121, 2163–2180.
- Goldstein, M.H.; Kiang, N.Y. Synchrony of Neural Activity in Electric Responses Evoked by Transient Acoustic Stimuli. J. Acoust. Soc. Am. 1958, 30, 107–114.
- Bourien, J.; Tang, Y.; Batrel, C.; Huet, A.; Lenoir, M.; Ladrech, S.; Desmadryl, G.; Nouvian, R.; Puel, J.-L.; Wang, J. Contribution of auditory nerve fibers to compound action potential of the auditory nerve. J. Neurophysiol. 2014, 112, 1025–1039.
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89.
- Olichon, A.; Guillou, E.; Delettre, C.; Landes, T.; Arnauné-Pelloquin, L.; Emorine, L.J.; Mils, V.; Daloyau, M.; Hamel, C.; Amati-Bonneau, P.; et al. Mitochondrial dynamics and disease, OPA1. Biochim. Biophys. Acta 2006, 1763, 500–509.
- Frezza, C.; Cipolat, S.; De Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189.
- Lodi, R.; Tonon, C.; Valentino, M.L.; Iotti, S.; Clementi, V.; Malucelli, E.; Barboni, P.; Longanesi, L.; Schimpf, S.; Wissinger, B.; et al. Deficit of in vivo mitochondrial ATP production in OPA1-related dominant optic atrophy. Ann. Neurol. 2004, 56, 719–723.
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ’plus’ phenotypes. Brain 2008, 131, 338–351.
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain A J. Neurol. 2010, 133, 771–786.
- Miyamoto, R.T.; Kirk, K.H.; Renshaw, J.; Hussain, D. Cochlear Implantation in Auditory Neuropathy. Laryngoscope 1999, 109, 181–185.
- Frewin, B.; Chung, M.; Donnelly, N. Bilateral cochlear implantation in Friedreich’s ataxia: A case study. Cochlear Implants Int. 2013, 14, 287–290.
- Rodríguez-Ballesteros, M.; Reynoso, R.; Olarte, M.; Villamar, M.; Morera, C.; Santarelli, R.; Arslan, E.; Medá, C.; Curet, C.; Völter, C.; et al. A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum. Mutat. 2008, 29, 823–831.
- Roux, I.; Safieddine, S.; Nouvian, R.; Grati, M.; Simmler, M.-C.; Bahloul, A.; Perfettini, I.; Le Gall, M.; Rostaing, P.; Hamard, G.; et al. Otoferlin, Defective in a Human Deafness Form, Is Essential for Exocytosis at the Auditory Ribbon Synapse. Cell 2006, 127, 277–289.
- Pangršič, T.; Lasarow, L.; Reuter, K.; Takago, H.; Schwander, M.; Riedel, D.; Frank, T.; Tarantino, L.M.; Bailey, J.S.; Strenzke, N.; et al. Hearing requires otoferlin-dependent efficient replenishment of synaptic vesicles in hair cells. Nat. Neurosci. 2010, 13, 869–876.
- Santarelli, R.; del Castillo, I.; Rodríguez-Ballesteros, M.; Scimemi, P.; Cama, E.; Arslan, E.; Starr, A. Abnormal Cochlear Potentials from Deaf Patients with Mutations in the Otoferlin Gene. J. Assoc. Res. Otolaryngol. 2009, 10, 545–556.
- Akil, O.; Dyka, F.; Calvet, C.; Emptoz, A.; Lahlou, G.; Nouaille, S.; de Monvel, J.B.; Hardelin, J.-P.; Hauswirth, W.W.; Avan, P.; et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc. Natl. Acad. Sci. USA 2019, 116, 4496–4501.
- Rankovic, V.; Vogl, C.; Dörje, N.M.; Bahader, I.; Duque-Afonso, C.J.; Thirumalai, A.; Weber, T.; Kusch, K.; Strenzke, N.; Moser, T. Overloaded Adeno-Associated Virus as a Novel Gene Therapeutic Tool for Otoferlin-Related Deafness. Front. Mol. Neurosci. 2021, 13, 253.