Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Agriculture, Dairy & Animal Science
The proteasome is responsible for selective degradation of most cellular proteins. Abundantly present in the cell, proteasomes not only diffuse in the cytoplasm and the nucleus but also associate with the chromatin, cytoskeleton, various membranes and membraneless organelles/condensates. How and why the proteasome gets to these specific subcellular compartments remains poorly understood, although increasing evidence supports the hypothesis that intracellular localization may have profound impacts on the activity, substrate accessibility and stability/integrity of the proteasome.
- proteasome
- membrane
- nucleus
- condensate
- ubiquitination
- myristoylation
1. Introduction
The 26S proteasome is situated at the core of the ubiquitin–proteasome system (UPS), responsible for selective degradation of the majority of cellular proteins in eukaryotes. For over three decades since its discovery, the proteasome has been thoroughly studied with regard to its composition, structure, activity, regulation and relation to health and disease. The fully assembled 26S proteasome holoenzyme consists of a 20S core particle (CP, formed by homologous α and β type subunits) and one or two 19S regulatory particles (RP, formed by six ATPase subunits called Rpt1-6, and thirteen non-ATPase subunits known as Rpns) [1,2,3,4,5,6,7,8,9,10]. Recent structural studies have significantly furthered our knowledge about how the proteasome recognizes and processes ubiquitinated substrates [6,11,12]. The success of proteasome inhibitors (e.g., Bortezomib/Velcade®) in treating multiple myeloma [13] has spurred intensive research on developing proteasome-targeting compounds for therapeutic uses toward cancer and autoimmune diseases, whereas (re-)activating the proteasome by small molecules has also emerged as an attractive strategy for alleviating symptoms associated with neurodegeneration and aging [14,15,16,17,18,19]. A better understanding of the function and regulation of the proteasome is of great biological and clinical importance.
As a soluble and highly abundant macromolecular complex [20,21,22], the proteasome resides in both the nucleus and cytoplasm of a cell and has been found associated with various subcellular structures, including the chromatin, cytoskeleton, nuclear envelope, plasma membrane, the cytosolic side of membrane-bound organelles and membraneless organelles/condensates (see below). Despite their pervasive presence, proteasomes are not evenly distributed in all cells. On a global scale, asymmetric cell division can lead to unequal inheritance of proteasomes between the daughter cells [23,24,25,26]. The specific subcellular localizations of proteasomes are often cell type- and growth status-dependent and dynamically regulated under both basal and stimulated/stress conditions [27,28,29]. A classic example is that in yeast, proteasomes are predominantly present in the nucleus of proliferating cells; but upon quiescence or carbon starvation, nuclear proteasomes are rapidly exported to the cytosol, where they are concentrated in a membraneless structure called proteasome storage granule (PSG) [30]. PSGs quickly resolve when yeast cells resume growth in nutrient-rich media and proteasomes re-gather in the nucleus. This reversible process is believed to protect the proteasome repertoire from autophagic degradation under stress conditions, while allowing them to regain function as soon as the stress is relieved [30,31].
Proteasomes also exist extracellularly. Original studies have shown that secreted proteasomes from ascidian sperms can digest vitelline coat proteins outside the egg and are required for egg penetration and fertilization [32,33,34]. Circulating proteasomes (c-proteasomes) were also found in humans around the same time [35], which has been confirmed by a series of subsequent studies (see reviews [36,37,38] and references therein). Present in the blood as well as other bodily fluids, these c-proteasomes are mostly in the form of 20S, probably due to the low-ATP extracellular environment that does not support RP–CP association [39,40]. Nonetheless, they are enzymatically active, and elevation of their levels is often correlated with either malignancy or tissue injury/damage, making them a promising biomarker for disease diagnosis [36,37,38]. How the proteasomes exit the cell remains a matter of debate, although a likely mechanism is via exosome-mediated non-conventional secretion [36,41]. The pathophysiological roles and regulatory mechanisms of extracellular proteasomes have yet to be fully understood.
Various mechanisms have been identified to target protein substrates to different subcellular regions for proteasomal degradation [42,43,44,45,46,47,48,49,50]. On the flip side, proteasomes should be available at the site of degradation or can be mobilized to meet the substrates. In addition to the examples introduced above, the dynamic localizations of the proteasome have been extensively studied (particularly in yeast) and summarized in a series of reviews [51,52,53,54,55]. Here, I will focus on the latest findings about nuclear-localized and membrane-associated proteasomes in mammalian cells and discuss the targeting mechanisms, biological functions, as well as regulations of proteasomes at these specific compartments.
2. Proteasomes at the Membranes
Membrane localization of the proteasome has been documented since the early 1990′s [116,117]. Numerous subsequent studies have documented proteasomes in close contact with nuclear envelope-ER [92,118,119,120,121,122,123,124], the Golgi apparatus [125,126], endosomes [127,128], plasma membrane [129,130], mitochondria [123,131,132,133,134,135,136] and so on. Proteasomes at the membranes are particularly important for organelle quality control processes, such as ER-associated degradation (ERAD), endosome and Golgi-associated degradation (EGAD) and mitophagy [120,125,126,137,138,139,140,141]. In addition, proteasomes located at neuronal synapses are also critical for neurotransmission and synaptic plasticity [142,143,144,145,146]. In these cases, the proteasome associates peripherally with the membrane by binding to membrane-resident proteins. This can occur directly between proteasome subunits and membrane proteins. Alternatively, proteasomes can be indirectly recruited to the membrane through binding to the ubiquitin moiety of modified membrane proteins, proteasome-interacting proteins (sometimes in concert with motor proteins and cytoskeleton) [147,148] or even RNAs that function as protein scaffolds [149]. Together, these represent the most common mode of proteasome–membrane interaction, while the proteasome can also locate to the membrane in two other ways, as elaborated below (Figure 1).
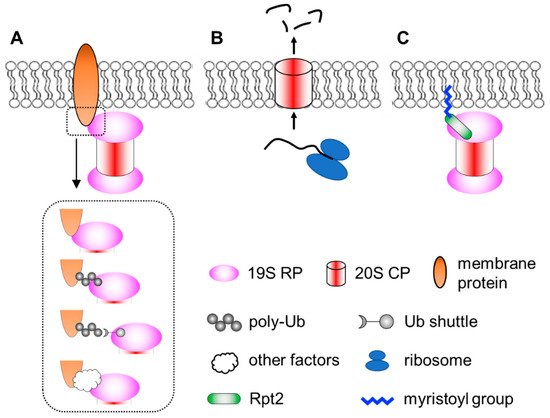
Figure 1. A simplified view of different modes of proteasome–membrane interaction. (A) In most cases, proteasomes attach to the membrane by directly or indirectly binding to resident membrane proteins, which may be modified by ubiquitination. (B) Neuronal membrane proteasomes (NMP) can degrade nascent, unfolded polypeptides. (C) Proteasomes tethered to the membrane via N-myristoylated Rpt2, which is evolutionarily conserved from yeast to human.
2.1. Neuronal Membrane Proteasomes
Ramachandran et al. reported a surprising type of membrane-associated proteasomes designated as the neuronal membrane proteasomes (NMPs) [150,151]. As the name suggests, these proteasomes are found at pre- and post-synaptic plasma membranes in neurons, which were confirmed by immunogold electron microscopy (IEM), surface biotinylation, immunofluorescence imaging with antibody feeding and proteinase protection assays. NMPs are thought to be comprised of the 20S CP only, since no 19S components (such as Rpt5 or Rpn1) were found by IEM in these particular membrane proteasomes. NMPs are capable of degrading newly synthesized polypeptides, which are still unfolded, to short peptides. More fascinatingly, the authors showed that these peptide products could exit the cells through NMPs and be released into the synaptic cleft to function as neurotransmitters. Therefore, NMPs function not only as a protein degrader but also a new form of membrane channel to mediate cell–cell communications [152]. Although these findings were very unique and intriguing, the molecular and biochemical details of the NMPs remain unclear. First, it is curious that the 20S CP, which is soluble and hydrophilic, could be fully embedded within the hydrophobic membrane. How is the CP targeted to the plasma membrane and how does it overcome the energy barrier to traverse the lipid bilayer? It was proposed that glycoproteins, such as GPM6, could facilitate this process [151], but a clear mechanistic explanation is still needed. Second, does the NMP exhibit any substrate selectivity? The proposed role of NMPs in cleaving nascent proteins suggests that substrate availability depends on localized protein synthesis by ribosomes in the vicinity [150]. However, if the NMP complex also contained auxiliary factors yet to be identified, it might recognize and process folded protein substrates as well. On the other hand, the recent discovery that the 20S CP can by itself degrade ubiquitinated proteins [153] also implies that NMPs may have a broader range of substrates. A following question is the molecular composition and regulatory mechanisms of the NMPs. Finally, what is the function of NMP in vivo? Additionally, how can we specifically maneuver it for research and therapeutic purposes without affecting the bulk of proteasomes inside the cell? Answering these questions will depend on new technical advances in imaging, chemical biology, proteomics, structural biology and genetic models, which makes it challenging but also rewarding at the same time.
2.2. Membrane Targeting of Proteasomes by N-Myristoylation
A third means of targeting the proteasome to the membrane is through lipid modification. N-myristoylation of the Rpt2 subunit has been observed by mass spectrometry in multiple species, ranging from yeast to plants to mammals [154,155,156,157,158,159,160,161]. Typically, N-myristoylation occurs co-translationally on nascent polypeptides still bound to the ribosome, where the 14-carbon saturated fatty acyl group is covalently linked to the second amino acid (almost always a Gly) after the initiator methionine is removed by methionyl aminopeptidase [162,163,164]. Notably, among all proteasome subunits of mammalian cells, Rpt2 is the only one that begins with Met-Gly, serving as the only site of the entire proteasome complex for N-myristoylation. This MG sequence of Rpt2 is strictly conserved from yeast to human, suggesting that Rpt2 is likely to be myristoylated in all species. In yeast, myristoylated Rpt2 has been shown to target proteasomes to the nuclear envelope, which is required for nuclear protein quality control [156,157]. Blocking this modification with the Rpt2-ΔG or Rpt2-G2A mutations causes mislocalization of nuclear proteasomes to the cytosol.
The role of Rpt2 myristoylation in higher organisms has not been rigorously investigated, despite Rpt2 being one of the most abundantly myristoylated proteins in human cells [161]. Our recent work demonstrated that wild-type human Rpt2 proficient for myristoylation was found at the plasma membrane, with some distribution at membrane-bound organelles as well. Membrane localization was abolished by the same ΔG/G2A mutations of human Rpt2. However, in stark contrast with results from yeast, loss of Rpt2 myristoylation in mammalian cells led to Rpt2 enrichment in the nucleus [84]. A serendipitous finding was that myristoylation-mediated membrane association is a prerequisite for Rpt2 phosphorylation at Tyr439 (Y439) by the tyrosine kinase Src, which itself is a well-established myristoylated protein tethered to the membrane [84,165]. Moreover, Rpt2-Y439 phosphorylation could be reversed by the phosphotyrosine phosphatase PTPN2 (also known as T cell PTP or TC-PTP). PTPN2 has multiple splicing isoforms. Rpt2-pY439 could only be dephosphorylated by the membrane-bound isoform of PTPN2 known as TC48, but not by the nuclear isoform TC45 [84]. Hence, the kinase, phosphatase and substrate are all placed in the same neighborhood confined by the membrane.
The biochemical consequence of Rpt2-Y439 phosphorylation is readily conceivable, as it is the very tyrosine residue within the highly conserved HbYX tail (hydrophobic residue—Tyr—any amino acid) of Rpt2 required for RP–CP association. Rpt2-Y439 is the most frequently detected pTyr site of all 19S subunits. The phosphorylation was seen in the developing rat brain but more evidently detected in cancer cells with hyperactive Src [84]. Src-mediated Rpt2-Y439 phosphorylation selectively inhibited the activity of membrane-associated proteasomes as demonstrated by a membrane-targeted reporter protein, MyrRpt2-GFPodc. On the contrary, the Src-specific inhibitor saracatinib/AZD0530 blocked Y439 phosphorylation and enhanced proteasomal degradation of membrane-bound substrates. Importantly, this seemed to be an integral part of the anti-cancer effects of saracatinib, since cancer cells expressing the nonphosphorylatable Y439F mutant were more resistant to this drug, both in vitro and in vivo [84]. Thus, reversible phosphorylation of Rpt2-Y439 provides a unique example of localized regulation of membrane-associated proteasomes.
3. Current Insights
Proteasome localization is highly dynamic within the cell and may be remarkably heterogeneous between cell types. This is an important basis of compartmentalized protein degradation that is widely conserved through evolution. Nonetheless, we have seen differences between yeast and mammalian cells where the behavior and fate of the proteasome are differentially controlled by specific factors. We are just beginning to get in-depth understanding of intracellular proteasome targeting and trafficking in higher organisms, and it remains a daunting mission to obtain a complete picture of localized function and regulation of the proteasome across different cell types, species and growth/stress conditions. A yet more challenging task would be to confirm these findings in vivo and to develop new tools for “site-specific” manipulation of proteasomes at any particular location in a cell.
A prerequisite for achieving these goals is a deeper and better characterization of proteasome composition, modification, interactome and its microenvironment within a cell. Researchers have been empowered by state-of-the-art techniques, including proximity labeling (e.g., BioID/TurboID, APEX, PUP-IT) [166,167,168], quantitative proteomics, super-resolution imaging and cryo-electron tomography (cryo-ET) [21,22,106,120,122] to probe and catalog the contents of proteasome-containing subcellular structures. Chemical biology approaches involving metabolic labeling, click chemistry, genetic code expansion and cross-linking mass spectrometry (XL-MS) have provided critical insights into proteasome modification and assembly [83,161,169,170,171]. Commonly used reporter proteins (e.g., GFPu, GFPodc, UbG76V-GFP, UBL-CP8-35) can be engineered to reflect local proteasome activity at defined compartments [84,108,172,173], while knock-in mice bearing fluorescence protein-tagged proteasome subunits would be valuable to monitor proteasome distribution and dynamics in vivo [174]. With classic yeast genetics and CRISPR screens, many more regulators of the proteasome are expected to be uncovered [93,114].
Finally, some further possibilities may be speculated. 1. In addition to the above discussed, what other mechanisms may be used for proteasome targeting? Can we alter proteasome localization (and function) pharmacologically, optically, mechanically, magnetically or acoustically [175]? Does lipid modification (i.e., N-myristoylation) promote exosomal secretion of the proteasome, as has been shown with palmitoylated ACE2 [176]? Can we design “Proteasome-TACs” that recruit proteasomes directly to the substrates (or vice versa) for therapeutic use? A recently discovered small circular RNA seemed to do precisely that [149]. Along this line, since proteasome subunits have been identified as RNA-binding proteins [177,178], can RNAs act as molecular tethers between the proteasome and chromatin or other proteins [179]? 2. As mentioned earlier, cancer cells show reduced SIPAN formation [109], and tyrosine phosphorylation of membrane-bound Rpt2 is relevant to the anti-cancer effect of saracatinib [84]. This makes one wonder whether proteasome (mis)localization can be considered as a biomarker for disease diagnosis and treatment. Moreover, it is unclear whether subcellular localization of the proteasome is altered in multiple myeloma patients after proteasome inhibitor treatment, or in patients with proteasome-associated autoinflammatory syndrome (PRAAS) who carry congenital mutations in proteasome genes [180,181]. Relocation of the proteasome may cause changes to the local proteome and rewire intracellular signaling, which may lead to a different kind of proteasome-oriented therapy.
This entry is adapted from the peer-reviewed paper 10.3390/biom12020229
This entry is offline, you can click here to edit this entry!