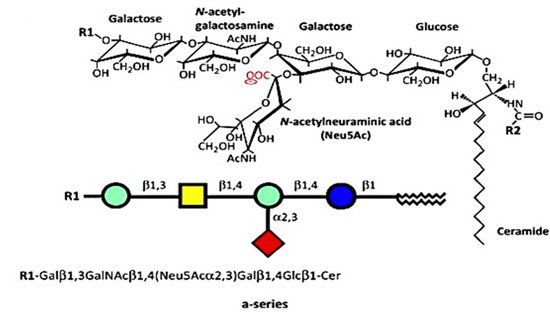
The numerous glycosphingolipids that occur in the nervous system and elsewhere are clearly involved in metabolic and pathological changes that accompany Parkinson’s disease, but one such molecule, GM1 ganglioside, has received focused attention from several workers for its prominent role in both the etiology and potential treatment of this neurodegenerative condition. GM1 is particularly abundant in neurons and is essential for their complex functioning (see below). The same may be said for ganglioside GD1a, which is identical in structure to GM1 but possesses an additional sialic acid attached to the terminal galactose of GM1. That terminal sialic acid is readily removed by NEU3 neuraminidase, which is situated close to GD1a in the neuronal membrane. This is why GD1a is considered to function as a reserve pool for GM1 as its primary function.
- Parkinson’s disease
- GM1
- ganglioside
- neuroprotection
1. Introduction
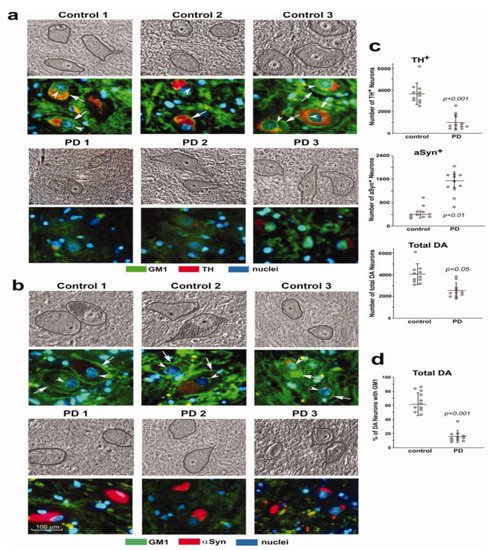
2. GM1 Deficiency Induces Parkinsonism in Mouse PD Model
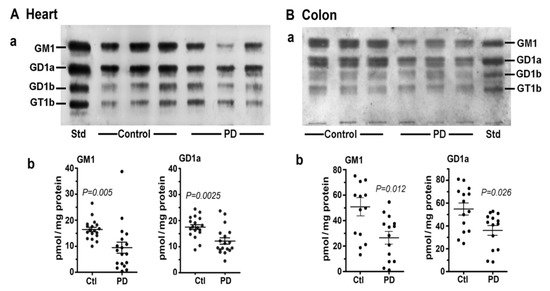
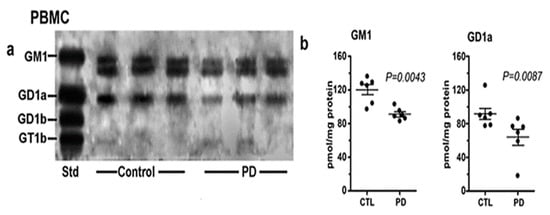
3. Systemic Deficiency of GM1 in PD Tissues
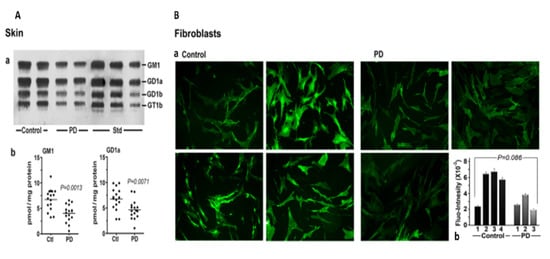
4. Cause of GM1 Deficiency
5. GM1 Decreases in the Periphery as well as Brain
6. GM1 and Neurotrophic Factors
7. Conclusions and Therapeutic Implications
This entry is adapted from the peer-reviewed paper 10.3390/biom12020173
References
- Piccinini, M.; Scandroglio, F.; Prioni, S.; Buccinnà, B.; Loberto, N.; Aureli, M.; Chigorno, V.; Lupino, E.; DeMarco, G.; Lomartire, A.; et al. Deregulated Sphingolipid Metabolism and Membrane Organization in Neurodegenerative Disorders. Mol. Neurobiol. 2010, 41, 314–340.
- Ledeen, R.W.; Wu, G. Gangliosides of the Nervous System. In Methods in Molecular Biology; Sonnino, S., Prinetti, A., Eds.; Elsevier: New York, NY, USA, 2018; Volume 1804, pp. 19–25.
- Miyagi, T.; Yamaguchi, K. Mammalian sialidases: Physiological and pathological roles in cellular functions. Glycobiology 2012, 22, 880–896.
- Ledeen, R.W.; Wu, G. The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem. Sci. 2015, 40, 407–418.
- Wu, G.; Lu, Z.-H.; Kulkarni, N.; Amin, R.; Ledeen, R.W. Mice Lacking Major Brain Gangliosides Develop Parkinsonism. Neurochem. Res. 2011, 36, 1706–1714.
- Wu, G.; Lu, Z.-H.; Kulkarni, N.; Ledeen, R.W. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J. Neurosci. Res. 2012, 90, 1997–2008.
- Hadaczek, P.; Wu, G.; Sharma, N.; Ciesielska, A.; Bankiewicz, K.; Davidow, A.L.; Lu, Z.H.; Forsayeth, J.; Ledeen, R.W. GDNF signaling implemented by GM1 ganglioside; failure in Parkinson’s disease and GM1-deficient murine model. Exp. Neurol. 2015, 263, 177–189.
- Ledeen, R.; Chowdhury, S.; Lu, Z.H.; Chakraborty, M.; Wu, G. Systemic deficiency of GM1 ganglioside in Parkinson’s disease tissues and its relation to the disease etiology. Glycoconj. J. 2022.
- Alselehdar, S.K.; Chakraborty, M.; Chowdhury, S.; Alcalay, R.N.; Surface, M.; Ledeen, R. Subnormal GM1 in PBMCs: Promise for Early Diagnosis of Parkinson’s Disease? Int. J. Mol. Sci. 2021, 22, 11522.
- Braak, H.; Del Tredici, K.; Rub, U.; De Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging. 2003, 24, 197–211.
- Braak, H.; De Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72.
- Pellicano, C.; Benincasa, D.; Pisani, V.; Buttarelli, F.R.; Giovannelli, M.; Pontieri, F.E. Prodromal non-motor symptoms of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 145–152.
- Halliday, G.; Barker, R.A.; Rowe, D.B. Non-dopamine Lesions in Parkinson’s Disease; Oxford University Press: Oxford, UK, 2013.
- Bonifati, V. Genetics of parkinsonism. Parkinsonism Relat. Disord. 2007, 13 (Suppl. 3), S233–S241.
- Svennerholm, L.; Boström, K.; Jungbjer, B.; Olsson, L. Membrane lipids of adult human brain: Lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J. Neurochem. 1994, 63, 1802–1811.
- Svennerholm, L.; Boström, K.; Fredman, P.; Månsson, J.-E.; Rosengren, B.; Rynmark, B.-M. Human brain gangliosides: Developmental changes from early fetal stage to advanced age. Biochimi. Biophys. Acta 1989, 1005, 109–117.
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; Consortium, I.P.s.D.G. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203.
- Guo, Y.L.; Duan, W.J.; Lu, D.H.; Ma, X.H.; Li, X.X.; Li, Z.; Bi, W.; Kurihara, H.; Liu, H.Z.; Li, Y.F.; et al. Autophagy-dependent removal of α-synuclein: A novel mechanism of GM1 ganglioside neuroprotection against Parkinson’s disease. Acta Pharmacol. Sin. 2021, 42, 518–528.
- Keshavarzian, A.; Engen, P.; Bonvegna, S.; Cilia, R. Chapter 11—The gut microbiome in Parkinson’s disease: A culprit or a bystander? In Progress in Brain Research; Björklund, A., Cenci, M.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 252, pp. 357–450.
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 2016, 32, 66–72.
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552.
- Chen, J.S.; Faller, D.V.; Spanjaard, R.A. Short-chain fatty acid inhibitors of histone deacetylases: Promising anticancer therapeutics? Curr. Cancer Drug Targets 2003, 3, 219–236.
- Tsai, Y.T.; Yu, R.K. Epigenetic activation of mouse ganglioside synthase genes: Implications for neurogenesis. J. Neurochem. 2014, 128, 101–110.
- Itokazu, Y.; Tsai, Y.T.; Yu, R.K. Epigenetic regulation of ganglioside expression in neural stem cells and neuronal cells. Glycoconj. J. 2017, 34, 749–756.
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164.
- Niimi, Y.; Mizutani, Y.; Akiyama, H.; Watanabe, H.; Shiroki, R.; Hirabayashi, Y.; Hoshinaga, K.; Mutoh, T. Cerebrospinal Fluid Profiles in Parkinson’s Disease: No Accumulation of Glucosylceramide, but Significant Downregulation of Active Complement C5 Fragment. J. Parkinsons Dis. 2021, 11, 221–232.
- Niimi, Y.; Ito, S.; Mizutani, Y.; Murate, K.; Shima, S.; Ueda, A.; Satake, W.; Hattori, N.; Toda, T.; Mutoh, T. Altered regulation of serum lysosomal acid hydrolase activities in Parkinson’s disease: A potential peripheral biomarker? Parkinsonism Relat. Disord. 2019, 61, 132–137.
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry 2007, 46, 1868–1877.
- Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE 2014, 9, e103727.
- Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 Ganglioside Modifies α-Synuclein Toxicity and is Neuroprotective in a Rat α-Synuclein Model of Parkinson’s Disease. Sci. Rep. 2019, 9, 8362.
- Ledeen, R.W.; Skrivanek, J.A.; Tirri, L.J.; Margolis, R.K.; Margolis, R.U. Gangliosides of the neuron: Localization and origin. Adv Exp. Med. Biol. 1976, 71, 83–103.
- Sonnino, S.; Ghidoni, R.; Fiorilli, A.; Venerando, B.; Tettamanti, G. Cytosolic gangliosides of rat brain: Their fractionation into protein-bound complexes of different ganglioside compositions. J. Neurosci. Res. 1984, 12, 193–204.
- Mutoh, T.; Tokuda, A.; Miyadai, T.; Hamaguchi, M.; Fujiki, N. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc. Natl. Acad. Sci. USA 1995, 92, 5087–5091.
- Pitto, J.; Mutoh, T.; Kuriyama, M.; Ferraretto, A.; Palestini, P.; Masserini, M. Influence of endogenous GM1 ganglioside on TrkB activity in cultured neurons. FEBS Lett. 1998, 439, 93–96.
- Pascual, A.; Hidalgo-Figueroa, M.; Piruat, J.I.; Pintado, C.O.; Gómez-Díaz, R.; López-Barneo, J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat. Neurosci. 2008, 11, 755–761.
- Chiricozzi, E.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Sonnino, S.; Mauri, L. GM1 Ganglioside Is A Key Factor in Maintaining the Mammalian Neuronal Functions Avoiding Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 868.
- Ledeen, R.W.; Wu, G. Gangliosides, α-Synuclein, and Parkinson’s Disease. In Progress in Molecular Biology and Translational Science; Schnaar, R.L., Lopez, P.H.H., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 156, pp. 435–454.
- Schneider, J.S.; Sendek, S.; Daskalakis, C.; Cambi, F. GM1 ganglioside in Parkinson’s disease: Results of a five year open study. J. Neurol. Sci. 2010, 292, 45–51.
- Schneider, J.S.; Gollomp, S.M.; Sendek, S.; Colcher, A.; Cambi, F.; Du, W. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J. Neurol. Sci. 2013, 324, 140–148.
- Wu, G.; Lu, Z.H.; Seo, J.H.; Alselehdar, S.K.; DeFrees, S.; Ledeen, R.W. Mice deficient in GM1 manifest both motor and non-motor symptoms of Parkinson’s disease; successful treatment with synthetic GM1 ganglioside. Exp. Neurol. 2020, 329, 113284.