Transcription and replication are the two most essential processes that a cell does with its DNA: they allow cells to express the genomic content that is required for their functions and to create a perfect copy of this genomic information to pass on to the daughter cells. Nevertheless, these two processes are in a constant ambivalent relationship. When transcription and replication occupy the same regions, there is the possibility of conflicts between transcription and replication as transcription can impair DNA replication progression leading to increased DNA damage. Nevertheless, DNA replication origins are preferentially located in open chromatin next to actively transcribed regions, meaning that the possibility of conflicts is potentially an accepted incident for cells. Data in the literature point both towards the existence or not of coordination between these two processes to avoid the danger of collisions.
- transcription
- DNA replication
- genome instability
- DNA damage
- G-MiDS
- transcription–replication collision
1. Complexity of the Transcription Process

2. Transcription-Induced Genome Instability
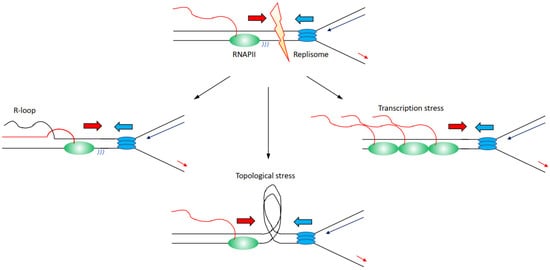
3. Co-Existence or Spatial–Temporal Separation between Transcription and Replication
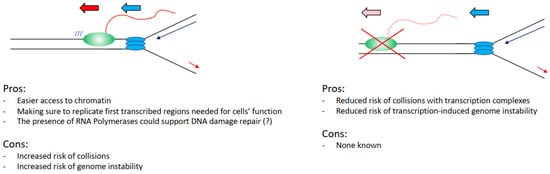
4. Pros and Cons from Coordinating Transcription and Replication
This entry is adapted from the peer-reviewed paper 10.3390/life12010108
References
- Stults, D.M.; Killen, M.W.; Pierce, H.H.; Pierce, A.J. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Biol. 2008, 18, 13–18.
- Pertea, M.; Shumate, A.; Pertea, G.; Varabyou, A.; Breitwieser, F.P.; Chang, Y.C.; Madugundu, A.K.; Pandey, A.; Salzberg, S.L. CHESS: A new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018, 19, 208.
- Dieci, G.; Fiorino, G.; Castelnuovo, M.; Teichmann, M.; Pagano, A. The expanding RNA polymerase III transcriptome. Trends Genet. 2007, 23, 614–622.
- Barski, A.; Chepelev, I.; Liko, D.; Cuddapah, S.; Fleming, A.B.; Birch, J.; Cui, K.; White, R.J.; Zhao, K. Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes. Nat. Struct. Mol. Biol. 2010, 17, 629–634.
- Canella, D.; Praz, V.; Reina, J.H.; Cousin, P.; Hernandez, N. Defining the RNA polymerase III transcriptome: Genome-wide localization of the RNA polymerase III transcription machinery in human cells. Genome Res. 2010, 20, 710–721.
- Raha, D.; Wang, Z.; Moqtaderi, Z.; Wu, L.; Zhong, G.; Gerstein, M.; Struhl, K.; Snyder, M. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc. Natl. Acad. Sci. USA 2010, 107, 3639–3644.
- Gerber, A.; Ito, K.; Chu, C.S.; Roeder, R.G. Gene-Specific control of tRNA expression by RNA Polymerase II. Mol. Cell 2020, 78, 765–778.
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA Polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65.
- Abraham, K.J.; Khosraviani, N.; Chan, J.N.Y.; Gorthi, A.; Samman, A.; Zhao, D.Y.; Wang, M.; Bokros, M.; Vidya, E.; Ostrowski, L.A.; et al. Nucleolar RNA polymerase II drives ribosome biogenesis. Nature 2020, 585, 298–302.
- Pennisi, E. Genomics. ENCODE project writes eulogy for junk DNA. Science 2012, 337, 1159–1161.
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108.
- Proudfoot, N.J. Transcriptional interference and termination between duplicated alpha-globin gene constructs suggests a novel mechanism for gene regulation. Nature 1986, 322, 562–565.
- Prescott, E.M.; Proudfoot, N.J. Transcriptional collision between convergent genes in budding yeast. Proc. Natl. Acad. Sci. USA 2002, 99, 8796–8801.
- Hobson, D.J.; Wei, W.; Steinmetz, L.M.; Svejstrup, J.Q. RNA polymerase II collision interrupts convergent transcription. Mol. Cell 2012, 48, 365–374.
- Yardimci, H.; Wang, X.; Loveland, A.B.; Tappin, I.; Rudner, D.Z.; Hurwitz, J.; van Oijen, A.M.; Walter, J.C. Bypass of a protein barrier by a replicative DNA helicase. Nature 2012, 492, 205–209.
- Hamperl, S.; Cimprich, K.A. Conflict resolution in the genome: How transcription and replication make it work. Cell 2016, 167, 1455–1467.
- Gomez-Gonzalez, B.; Aguilera, A. Transcription-mediated replication hindrance: A major driver of genome instability. Genes Dev. 2019, 33, 1008–1026.
- Pomerantz, R.T.; O’Donnell, M. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature 2008, 456, 762–766.
- Pomerantz, R.T.; O’Donnell, M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science 2010, 327, 590–592.
- Merrikh, H.; Machón, C.; Grainger, W.H.; Grossman, A.D.; Soultanas, P. Co-directional replication-transcription conflicts lead to replication restart. Nature 2011, 470, 554–557.
- Prado, F.; Aguilera, A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005, 24, 1267–1276.
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-replication conflict orientation modulates R-Loop levels and activates distinct DNA damage responses. Cell 2017, 170, 774–786.
- Wang, J.D.; Berkman, M.B.; Grossman, A.D. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2007, 104, 5608–5613.
- Mirkin, E.V.; Mirkin, S.M. Mechanisms of transcription-replication collisions in bacteria. Mol. Cell Biol. 2005, 25, 888–895.
- Dutta, D.; Shatalin, K.; Epshtein, V.; Gottesman, M.E.; Nudler, E. Linking RNA polymerase backtracking to genome instability in E. coli. Cell 2011, 146, 533–543.
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721.
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 2009, 35, 228–239.
- Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439.
- Castellano-Pozo, M.; Santos-Pereira, J.M.; Rondón, A.G.; Barroso, S.; Andújar, E.; Pérez-Alegre, M.; García-Muse, T.; Aguilera, A. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol. Cell 2013, 52, 583–590.
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLOS Genet. 2014, 10, e1004318.
- Bermejo, R.; Capra, T.; Gonzalez-Huici, V.; Fachinetti, D.; Cocito, A.; Natoli, G.; Katou, Y.; Mori, H.; Kurokawa, K.; Shirahige, K.; et al. Genome-organizing factors Top2 and Hmo1 prevent chromosome fragility at sites of S phase transcription. Cell 2009, 138, 870–884.
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324.
- Promonet, A.; Padioleau, I.; Liu, Y.; Sanz, L.; Biernacka, A.; Schmitz, A.L.; Skrzypczak, M.; Sarrazin, A.; Mettling, C.; Rowicka, M.; et al. Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat. Commun. 2020, 11, 3940.
- Saponaro, M.; Kantidakis, T.; Mitter, R.; Kelly, G.P.; Heron, M.; Williams, H.; Söding, J.; Stewart, A.; Svejstrup, J.Q. RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell 2014, 157, 1037–1049.
- Poetch, A.R.; Boulton, S.J.; Luscombe, N.M. Genomic landscape of oxidative DNA damage and repair reveals regioselective protection from mutagenesis. Genome Biol. 2018, 19, 215.
- Wu, W.; Hill, S.E.; Nathan, W.J.; Paiano, J.; Callen, E.; Wang, D.; Shinoda, K.; van Wietmarschen, N.; Colón-Mercado, J.M.; Zong, D.; et al. Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 2021, 593, 440–444.
- Jiang, Y.; Li, W.; Lindsey-Boltz, L.A.; Yang, Y.; Li, Y.; Sancar, A. Super hotspots and super coldspots in the repair of UV-induced DNA damage in the human genome. JBC 2021, 296, 100581.
- Marinello, J.; Bertoncini, S.; Aloisi, I.; Cristini, A.; Malagoli Tagliazucchi, G.; Forcato, M.; Sordet, O.; Capranico, G. Dynamic effects of topoisomerase I inhibition on R-Loops and short transcripts at active promoters. PLoS ONE 2016, 11, e0147053.
- Zatreanu, D.; Han, Z.; Mitter, R.; Tumini, E.; Williams, H.; Gregersen, L.; Dirac-Svejstrup, A.B.; Roma, S.; Stewart, A.; Aguilera, A.; et al. Elongation factor TFIIS prevents transcription stress and R-Loop accumulation to maintain genome stability. Mol. Cell 2019, 76, 57–69.
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142.
- Debacker, K.; Frank Kooy, R. Fragile sites and human disease. Hum. Mol. Genet. 2007, 16, R150–R158.
- Debatisse, M.; Le Tallec, B.; Letessier, A.; Dutrillaux, B.; Brison, O. Common fragile sites: Mechanisms of instability revisited. Trends Genet. 2012, 28, 22–32.
- Bignell, G.R.; Greenman, C.D.; Davies, H.; Butler, A.P.; Edkins, S.; Andrews, J.M.; Buck, G.; Chen, L.; Beare, D.; Latimer, C.; et al. Signatures of mutation and selection in the cancer genome. Nature 2010, 463, 893–898.
- Hazan, I.; Hofman, T.G.; Aqeilan, R.i. Tumor suppressor genes within common fragile sites are active players in the DNA damage response. PLOS Genet. 2016, 12, e1006436.
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789.
- Pirzio, L.M.; Pichierri, P.; Bignami, M.; Franchitto, A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J. Cell Biol. 2008, 180, 305–314.
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123.
- Bergoglio, V.; Boyer, A.S.; Walsh, E.; Naim, V.; Legube, G.; Lee, M.Y.; Rey, L.; Rosselli, F.; Cazaux, C.; Eckert, K.A.; et al. DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J. Cell Biol. 2013, 201, 395–408.
- Le Tallec, B.; Millot, G.A.; Blin, M.E.; Brison, O.; Dutrillaux, B.; Debatisse, M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 2013, 4, 420–428.
- Brison, O.; El-Hilali, S.; Azar, D.; Koundrioukoff, S.; Schmidt, M.; Nähse, V.; Jaszczyszyn, Y.; Lachages, A.M.; Dutrillaux, B.; Thermes, C.; et al. Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide. Nat. Commun. 2019, 10, 5693.
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632.
- Howard, A.; Pelc, S.R. Synthesis of deoxyribonucleic acid in normal and irradiated cells and its relation to chromosome breakage. Heredity 1953, 6, 261–273.
- Minocherhomji, S.; Ying, S.; Bjerregaard, V.A.; Bursomanno, S.; Aleliunaite, A.; Wu, W.; Mankouri, H.W.; Shen, H.; Liu, Y.; Hickson, I.D. Replication stress activates DNA repair synthesis in mitosis. Nature 2015, 528, 286–290.
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 2016, 64, 1117–1126.
- Maya-Mendoza, A.; Moudry, P.; Merchut-Maya, J.M.; Lee, M.; Strauss, R.; Bartek, J. High speed of fork progression induces DNA replication stress and genomic instability. Nature 2018, 559, 279–284.
- Klein, J.; Grummt, I. Cell cycle-dependent regulation of RNA polymerase I transcription: The nucleolar transcription factor UBF is inactive in mitosis and early G1. Proc. Natl. Acad. Sci. USA 1999, 96, 6096–6101.
- Brewer, B.J.; Fangman, W.L. A replication fork barrier at the 3′ end of yeast ribosomal RNA genes. Cell 1988, 55, 637–643.
- Akamatsu, Y.; Kobayashi, T. The human RNA Polymerase I transcription terminator complex acts as a replication fork barrier that coordinates the progress of replication with rRNA transcription activity. Mol. Cell Biol. 2015, 35, 1871–1881.
- Warmerdam, D.O.; Wolthuis, R.M.F. Keeping ribosomal DNA intact: A repeating challenge. Chromosome Res. 2019, 27, 57–72.
- White, R.J.; Gottlieb, T.M.; Downes, C.S.; Jackson, S.P. Cell cycle regulation of RNA polymerase III transcription. Mol. Cell Biol. 1995, 15, 6653–6662.
- Scott, P.H.; Cairns, C.A.; Sutcliffe, J.E.; Alzuherri, H.M.; McLees, A.; Winter, A.G.; White, R.J. Regulation of RNA polymerase III transcription during cell cycle entry. JBC 2001, 276, 1005–1014.
- Gottesfeld, J.M.; Wolf, V.J.; Dang, T.; Forbes, D.J.; Hartl, P. Mitotic repression of RNA polymerase III transcription in vitro mediated by phosphorylation of a TFIIIB component. Science 1994, 263, 81–84.
- Azvolinsky, A.; Giresi, P.G.; Lieb, J.D.; Zakian, V.A. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell 2009, 34, 722–734.
- Liang, K.; Woodfin, A.R.; Slaughter, B.D.; Unruh, J.R.; Box, A.C.; Rickels, R.A.; Gao, X.; Haug, J.S.; Jaspersen, S.L.; Shilatifard, A. Mitotic transcriptional activation: Clearance of actively engaged Pol II via transcriptional elongation control in mitosis. Mol. Cell 2015, 60, 435–445.
- Palozola, K.C.; Donahue, G.; Liu, H.; Grant, G.R.; Becker, J.S.; Cote, A.; Yu, H.; Raj, A.; Zaret, K.S. Mitotic transcription and waves of gene reactivation during mitotic exit. Science 2017, 358, 119–122.
- van der Meijden, C.M.; Lapointe, D.S.; Luong, M.X.; Peric-Hupkes, D.; Cho, B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Gene profiling of cell cycle progression through S-phase reveals sequential expression of genes required for DNA replication and nucleosome assembly. Cancer Res. 2002, 62, 3233–3243.
- Schübeler, D.; Scalzo, D.; Kooperberg, C.; van Steensel, B.; Delrow, J.; Groudine, M. Genome-wide DNA replication profile for Drosophila melanogaster: A link between transcription and replication timing. Nat. Genet. 2002, 32, 438–442.
- Woodfine, K.; Fiegler, H.; Beare, D.M.; Collins, J.E.; McCann, O.T.; Young, B.D.; Debernardi, S.; Mott, R.; Dunham, I.; Carter, N.P. Replication timing of the human genome. Hum. Mol. Genet. 2004, 13, 191–202.
- Hansen, R.S.; Thomas, S.; Sandstrom, R.; Canfield, T.K.; Thurman, R.E.; Weaver, M.; Dorschner, M.O.; Gartler, S.M.; Stamatoyannopoulos, J.A. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. USA 2010, 107, 139–144.
- Karnani, N.; Taylor, C.M.; Malhotra, A.; Dutta, A. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol. Biol. Cell 2010, 21, 393–404.
- Dellino, G.I.; Cittaro, D.; Piccioni, R.; Luzi, L.; Banfi, S.; Segalla, S.; Cesaroni, M.; Mendoza-Maldonado, R.; Giacca, M.; Pelicci, P.G. Genome-wide mapping of human DNA-replication origins: Levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res. 2013, 23, 1–11.
- Petryk, N.; Kahli, M.; d’Aubenton-Carafa, Y.; Jaszczyszyn, Y.; Shen, Y.; Silvain, M.; Thermes, C.; Chen, C.L.; Hyrien, O. Replication landscape of the human genome. Nat. Commun. 2016, 7, 10208.
- Chen, Y.H.; Keegan, S.; Kahli, M.; Tonzi, P.; Fenyö, D.; Huang, T.T.; Smith, D.J. Transcription shapes DNA replication initiation and termination in human cells. Nat. Struct. Mol. Biol. 2019, 26, 67–77.
- Wei, X.; Samarabandu, J.; Devdhar, R.S.; Siegel, A.J.; Acharya, R.; Berezney, R. Segregation of transcription and replication sites into higher order domains. Science 1998, 281, 1502–1506.
- Hassan, A.B.; Errington, R.J.; White, N.S.; Jackson, D.A.; Cook, P.R. Replication and transcription sites are colocalized in human cells. J. Cell Sci. 1994, 107, 425–434.
- Meryet-Figuiere, M.; Alaei-Mahabadi, B.; Ali, M.M.; Mitra, S.; Subhash, S.; Pandey, G.K.; Larsson, E.; Kanduri, C. Temporal separation of replication and transcription during S-phase progression. Cell Cycle 2014, 13, 3241–3248.
- Wang, J.; Rojas, P.; Mao, J.; Mustè Sadurnì, M.; Garnier, O.; Xiao, S.; Higgs, M.R.; Garcia, P.; Saponaro, M. Persistence of RNA transcription during DNA replication delays duplication of transcription start sites until G2/M. Cell Rep. 2021, 34, 108759.
- St Germain, C.P.; Zhao, H.; Sinha, V.; Sanz, L.A.; Chedin, F.; Barlow, J.H. Genomic patterns of transcription-replication interactions in mouse primary B cells. Biorxiv 2021.
- Adar, S.; Hu, J.; Lieb, J.D.; Sancar, A. Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E2124–E2133.
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Cell Mol. Biol. 2014, 15, 465–481.
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374.
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive mapping of histone modifications at DNA double-strand breaks deciphers repair pathway chromatin signatures. Mol. Cell 2018, 72, 250–262.
- Pryde, F.; Khalili, S.; Robertson, K.; Selfridge, J.; Ritchie, A.M.; Melton, D.W.; Jullien, D.; Adachi, Y. 53BP1 exchanges slowly at the sites of DNA damage and appears to require RNA for its association with chromatin. J. Cell Sci. 2005, 118, 2043–2055.
- Francia, S.; Michelini, F.; Saxena, A.; Tang, D.; de Hoon, M.; Anelli, V.; Mione, M.; Carninci, P.; d’Adda di Fagagna, F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012, 488, 231–235.
- Pessina, F.; Giavazzi, F.; Yin, Y.; Gioia, U.; Vitelli, V.; Galbiati, A.; Barozzi, S.; Garre, M.; Oldani, A.; Flaus, A.; et al. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat. Cell Biol. 2019, 21, 1286–1299.
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 2019, 38, e101379.
- Michelini, F.; Pitchiaya, S.; Vitelli, V.; Sharma, S.; Gioia, U.; Pessina, F.; Cabrini, M.; Wang, Y.; Capozzo, I.; Iannelli, F.; et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 2017, 19, 1400–1411.
- Sharma, S.; Anand, R.; Zhang, X.; Francia, S.; Michelini, F.; Galbiati, A.; Williams, H.; Ronato, D.A.; Masson, J.Y.; Rothenberg, E.; et al. MRE11-RAD50-NBS1 complex is sufficient to promote transcription by RNA polymerase II at double-strand breaks by melting DNA ends. Cell Rep. 2021, 34, 108565.
- Ohle, C.; Tesorero, R.; Schermann, G.; Dobrev, N.; Sinning, I.; Fischer, T. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell 2016, 167, 1001–1013.
- Yasuhara, T.; Kato, R.; Hagiwara, Y.; Shiotani, B.; Yamauchi, M.; Nakada, S.; Shibata, A.; Miyagawa, K. Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell 2018, 175, 558–570.
- Cohen, S.; Puget, N.; Lin, Y.L.; Clouaire, T.; Aguirrebengoa, M.; Rocher, V.; Pasero, P.; Canitrot, Y.; Legube, G. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018, 9, 533.
- D’Alessandro, G.; Whelan, D.R.; Howard, S.M.; Vitelli, V.; Renaudin, X.; Adamowicz, M.; Iannelli, F.; Jones-Weinert, C.W.; Lee, M.; Matti, V.; et al. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat. Commun. 2018, 9, 5376.
- Aguilera, A.; Gomez-Gonzalez, B. DNA-RNA hybrids: The risks of DNA breakage during transcription. NSMB 2017, 24, 439–443.
- Marnef, A.; Legube, G. R-loops as Janus-faced modulators of DNA repair. Nat. Cell Biol. 2021, 23, 305–313.
- Rawal, C.C.; Zardoni, L.; Di Terlizzi, M.; Galati, E.; Brambati, A.; Lazzaro, F.; Liberi, G.; Pellicioli, A. Senataxin ortholog Sen1 limits DNA:RNA hybrid accumulation at DNA double-strand breaks to control end resection and repair fidelity. Cell Rep. 2020, 31, 107603.
- Ortega, P.; Mérida-Cerro, J.A.; Rondón, A.G.; Gómez-González, B.; Aguilera, A. DNA-RNA hybrids at DSBs interfere with repair by homologous recombination. Elife 2021, 10, e69881.