Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
|
Neurosciences
The PD model flies based on DJ-1β inactivation exhibited protein metabolism alterations, a shift from the tricarboxylic acid cycle to glycolytic pathway to obtain ATP, together with an increase in the expression of some urea cycle enzymes. Thus, these metabolic changes could contribute to PD pathogenesis and might constitute possible therapeutic targets and/or biomarkers for this disease.
- Drosophila
- DJ-1
- Parkinson’s disease
- metabolomics
- NMR spectroscopy
1. Introduction
Parkinson’s disease (PD) is the most common motor disorder, affecting more than 1% of the population over 60 years. PD is caused by the selective and progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc), which results in striatal dopamine deficiency and leads to disturbances in motor, autonomic and psychiatric functions [1][2][3]. DA neuron loss is accompanied, in some cases, by the presence of α-synuclein aggregates in the surviving neurons, known as Lewy bodies [1][3]. The exact mechanism behind DA neurodegeneration in PD is still unclear, most likely a combination of genetic predisposition and environmental factors [1][4]. Indeed, multiple pathways and mechanisms seem to participate in PD pathogenesis, such as the accumulation of misfolded protein aggregates, mitochondrial dysfunction, increased oxidative stress (OS), energy failure, neuroinflammation, and genetic mutations [5]. Interestingly, recent studies have shown that bioenergetic alterations may play a key role in PD neuropathology. Concretely, an increase in the glycolytic rate was observed in PD models, suggesting that there is a link between glucose metabolism, cellular bioenergetics, redox homeostasis and neuronal death [6][7].
Most PD cases are idiopathic (iPD), whose etiology is multifactorial. However, 5–10% of PD patients suffer from monogenic forms of the disease caused by highly penetrant mutations that are family-linked [1][4]. In recent years, mutations in several genes were associated with these familial forms of PD (fPD), allowing the discovery of different mechanisms underlying PD pathogenesis [4][8]. Indeed, some of the proteins encoded by these genes are involved in a set of molecular pathways that upon perturbation trigger a neuropathology that resembles or is clinically indistinguishable from iPD, except for the age at onset [9]. Among them, DJ-1 is a causative gene for fPD [10] that was initially described as an oncogene. Nevertheless, other functions were ascribed to the DJ-1 protein such as transcriptional regulation, chaperone and protease activity, scavenger of reactive oxygen species (ROS) or mitochondrial homeostasis [11][12][13]. Remarkably, an over-oxidized and inactive form of the DJ-1 protein was found in brains of iPD individuals, suggesting that it may play a central role in the development of the disease [14]. Therefore, results obtained in PD models with impaired DJ-1 function could be also applicable to human iPD forms.
Currently, PD diagnosis is limited and based on the detection of classic motor symptoms that appear when about 60–80% of SNpc DA neurons are lost, after many years of ongoing disease [15]. Thus, it is an urgent need to find simple, useful and low-cost biomarkers for an early PD diagnosis, in order to address its progression in the initial stages of the disease [16]. Biomarkers are also needed for distinguishing different PD types, predicting the course of the disease, or monitoring the effect of disease-modifying therapies [17][18]. Metabolomic approaches allow the analysis of a great number of low-molecular-weight molecules, offering an overview of the molecular complexity of a biological system and the metabolic pathways that can be altered in a pathological state [19][20]. Therefore, metabolomic studies are being carried out to identify new biomarkers in PD and other NDs [21][22], as well as therapeutic targets and metabolic alterations implicated in PD pathogenesis [22].
Drosophila melanogaster has emerged as an important model organism in the study of PD physiopathology. In this scenario, several genetic-based and chemically induced PD models were developed in Drosophila [23][24]. Among them, flies harboring mutations in DJ-1β (ortholog of DJ-1 human gene) present typical PD phenotypes, such as motor impairment, and increased levels of OS markers [25][26][27]. In addition, DJ-1β mutants exhibit an increased activity of key glycolytic enzymes [6]. Given the great complexity and heterogeneity in the metabolome of PD human samples, as well as their limited availability, models in simpler organisms such as Drosophila could be very useful for studying PD-associated, metabolome-wide alterations [28][29][30]. In fact, metabolomic studies were conducted in different fly models of other NDs. For instance, a metabolomic analysis of a Drosophila model of Charcot-Marie-Tooth disease based on GDAP1 deficiency allowed the involvement of insulin signaling in the characteristic neuromuscular degeneration of this disease to be identified [31]. Moreover, a transgenic Drosophila model of Huntington’s disease was used to discover metabolic perturbations at two stages of the disease [32]. Regarding PD, Shukla et al. [20] performed a metabolomic analysis in a paraquat-induced Drosophila model of iPD, in which altered levels of amino acids, lipids and carbohydrates were identified [20].
2. Current Insights
PD is an incurable and complex disease in which the mechanisms that cause DA neuronal death are not yet clear [1][4]. Some authors described a link between redox imbalance, energy failure and metabolic disturbances in PD, leading to the idea that it might be considered a metabolic disease [6][7]. In this study, we used NMR spectroscopy for metabolomic profiling in DJ-1β mutants and control flies of different ages. The aim of this study was to detect metabolic alterations that might be relevant in PD physiopathology, as well as identify potential therapeutic targets and biomarkers. To date, several metabolomic analyses were performed in the plasma, cerebrospinal fluid or blood of PD patients [33][34][35][36][37][38][39]. However, human samples are limited to specific tissues and do not allow for a complete examination of the disease [28][29][30]. To circumvent this limitation, we analyzed metabolites in the whole body of PD model flies. This experimental approach allows a complete panorama of PD-associated metabolic defects to be obtained, which are not restricted to the brain and can influence the physiology of the entire fly [32]. To date, only one similar study has been performed in another Drosophila PDf model based on PINK1 deficiency [40]. Our results confirm that several metabolic pathways, such as amino acid catabolism/anabolism, glycolysis, the TCA cycle, or the UC cycle, might be altered in PD.
Neurons require high levels of energy, which is mainly produced by mitochondria. However, mitochondria become less efficient with age as well as in several NDs, such as PD, thus leading to a reduction in ATP levels, while ROS production increases [41]. In this scenario, amino acids play an important role in the brain, since some of them can be metabolized to fuel cellular energetics when there is insufficient energy [42]. The amino acid profile observed in 1-day-old and 15-day-old DJ-1β mutants is consistent with amino acids anabolism/catabolism imbalance in PD model flies, especially in younger flies (Figure 1). Interestingly, an increase in amino acid catabolism, through transformation to pyruvate and acetyl-CoA or other TCA cycle intermediates to produce energy [43], could compensate ATP deficit due to impaired mitochondrial function. Moreover, the reduction in amino acid levels could be also consistent with impairment in protein synthesis in muscles, which could exacerbate motor symptoms in PD patients [44].
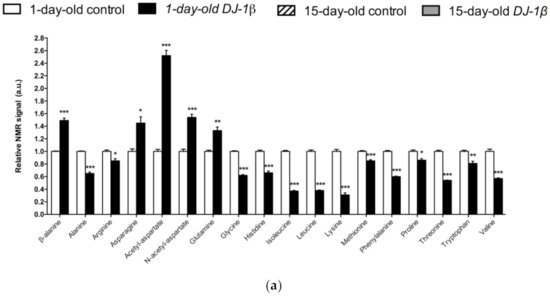
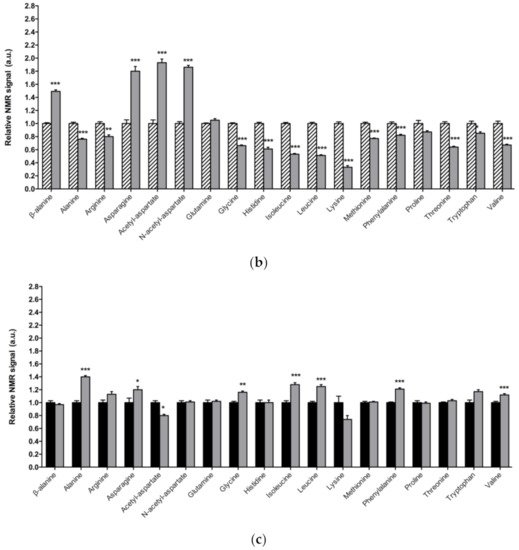
Figure 1. Alterations in amino acid content in PD model flies. Relative NMR signals of amino acids between (a) 1-day-old DJ-1β mutant and control flies, (b) 15-day-old DJ-1β mutant and control flies, and (c) 1-day-old and 15-day-old DJ-1β mutant flies. In all cases, error bars show s.d. from twelve independent samples (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Among the amino acids whose levels are altered in PD model flies compared to controls, we found BCAAs. BCAAs metabolism/degradation is closely related to carbon metabolism, in particular to glycolysis and the TCA cycle, since BCAAs can be transformed to succinyl-CoA that fuels TCA cycle [43]. PD model flies exhibit decreased levels of BCAAs probably due to its degradation to produce energy. A similar situation was found in a metabolomic analysis performed in sebum samples of PD patients [45], thus supporting the validity of the results obtained in our Drosophila PD model. Interestingly, a clinical trial (NCT01662414) was performed in PD patients to investigate the effect of whey protein supplementation, an important source of BCAAs. The results showed that this supplementation was able to increase BCAAs and essential amino acid levels, as well as reduced glutathione levels, a key antioxidant that prevents the oxidative damage of DA neurons [46]. In relation to this, a recent study showed that whey protein supplementation improved motor symptoms in PD patients due to an increase in muscle regeneration [47]. Our results indicate that BCAAs metabolism seems to be decreased in 15-day-old compared to 1-day-old DJ-1β mutant flies. This decrease, especially in aged flies, could enhance PD-related phenotypes, as shown in PD patients [46]. Therefore, BCAAs metabolism could constitute a possible therapeutic target to investigate new PD treatments. Levels of other proteinogenic amino acids are also reduced in DJ-1β mutant flies. For example, they present decreased tryptophan levels, whose metabolism is related to neurodegeneration in PD patients at early stages [48][43][49]. Another proteinogenic amino acid affected in DJ-1β mutants is glycine, a small amino acid with neuroprotective effects in neuroinflammation, ROS-related damage, and synaptic dysfunction through JNK signaling inactivation [50]. It is likely that a decrease in glycine levels in DJ-1β mutants could be contributing to PD pathology. Moreover, there is a significant change in glycine levels when comparing metabolomes of 1-day-old to 15-day-old mutant flies, thus suggesting that this amino acid could be a possible biomarker of PD progression.
PD model flies also exhibit significant increased levels of some non-proteinogenic amino acids compared to controls such as β-alanine. This result could indicate an increase in pyrimidine degradation, as β-alanine is the final product of this pathway. Similar results were found in pink1 mutant flies, another Drosophila fPD model [40]. Moreover, an increase in β-alanine levels causes taurine depletion, which was related to nerve degeneration [51]. Therefore, alterations in β-alanine metabolism and related pathways could be contributing to PD physiopathology. On the other hand, N-acetyl-aspartate is the most abundant non-proteinogenic amino acid in the central nervous system and a marker of neuronal integrity. It is synthetized in the mitochondria from aspartate and acetyl-CoA, and is then transported to the cytosol, where it is metabolized into aspartate and acetate. Its synthesis increases in absence of ATP due to the use of acetate as a source of energy [52]. We found that PD model flies showed an important increase in N-acetyl-aspartate (Figure 1), which could reflect a deficiency in its metabolism, since acetate levels were also decreased in 15-day-old DJ-1β mutants compared to control flies of the same age (Table S2).
Alterations in energy metabolism and glucose uptake are associated with the physiopathology of several NDs including PD [6][7][42]. Glucose is the main source of energy in the brain and the regulation of its metabolism is critical for brain physiology [41]. This is consistent with the observation that DJ-1β mutants show a dysregulation of carbohydrates metabolism (Figure 2). Supporting this, a decrease in glucose levels in the iPSC-derived DA neurons mutant for PARK2 was previously observed [53]. In fact, glucose is the sole substrate that can supply the rapid energy demand of neuronal cells through glycolysis [42][54]. Therefore, an increase in glucose consumption through this pathway to restore ATP levels could explain its reduction in our PD model flies [6][55]. Glycolysis is an ATP-producing pathway that could provide considerable amounts of ATP in a less efficient manner than oxidative phosphorylation to support the acute energy demands in neurons [54][56]. DJ-1 was described as participating in the activity of complex I of ETC binding and stabilizing it [13]. Accordingly, TCA cycle reduction and a higher NADH/NAD+ ratio in the PD model flies suggest the existence of an alteration of this complex and mitochondrial dysfunction, which was also observed in a MPTP-induced PD cell model [57]. Thus, our results show that a shift from TCA cycle to glycolysis is produced in PD model flies [6][58][59]. In addition, it was reported that a reduction in Aco activity caused neurotoxicity in mesencephalic rat cultures, due to the loss of its activity as ROS and an iron biosensor [60][61]. Moreover, Aco mutant flies exhibited a reduced locomotor activity, shortened lifespan, and increased cell death in the developing brain, as well as glycolysis and TCA cycle disturbances that led to decreased ATP levels [60]. Some of the phenotypes are similar to those observed in DJ-1β mutant flies [6][26]. On the other hand, the reduction in SDH activity leads to the activation of the mammalian target of rapamycin (mTOR) and the sterol regulatory element binding protein (SREBP), which contribute to lipid accumulation in neurons, as observed in several NDs, and produce excitotoxicity, thus participating in PD pathogenesis and development [62][63]. In addition, the finding of reduced malate levels in PD model flies is also noteworthy. Malate is an intermediate of the TCA cycle that is metabolized from fumarate by the fumarate hydratase enzyme. Interestingly, it was shown that the expression of fumarate hydratase was reduced in the DA neurons of the SNpc in the brains of iPD patients [64]. Thus, our results and these observations suggest that a decrease in the activity of both enzymes might be relevant to PD pathogenesis.
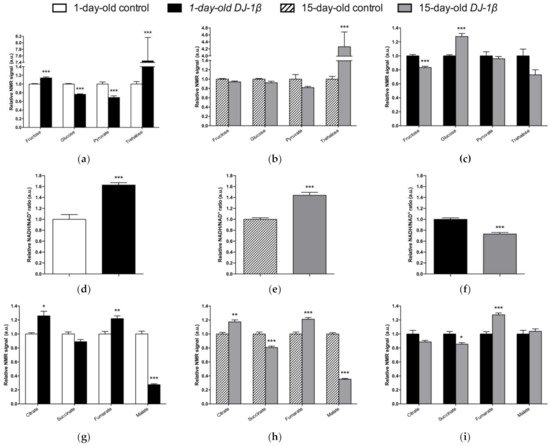
Figure 2. Alterations in carbohydrate content in DJ-1β mutant flies. Relative NMR signals of selected carbohydrates in between (a) 1-day-old DJ-1β mutant and control flies, (b) 15-day-old DJ-1β mutant and control flies, and (c) 1-day-old and 15-day-old DJ-1β mutant flies. Relative NADH/NAD+ ratio in (d) 1-day-old DJ-1β mutant and control flies, (e) 15-day-old DJ-1β mutant and control flies, and (f) 1-day-old and 15-day-old DJ-1β mutant flies. Relative NMR signals of TCA cycle intermediates comparing (g) 1-day-old DJ-1β mutant and control flies, (h) 15-day-old DJ-1β mutant and control flies, and (i) 1-day-old and 15-day-old DJ-1β mutant flies. In all cases, error bars show s.d. from twelve independent samples (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Although TCA cycle activity is decreased in DJ-1β mutant flies, there is an increase in some pathway intermediates such as fumarate (Figure 2g,h). TCA cycle metabolites can be produced by other pathways, as could be happening with fumarate and the UC [65]. UC is responsible for the excretion of the nitrogen that cannot be used in amino acid metabolism, and is related to the TCA cycle by fumarate, which is transported into the mitochondria, where it can be used as a substrate of TCA cycle [65][42]. The enhanced expression of arg and Argl in PD model flies could lead to a general increase in UC activity and to higher fumarate levels, as observed in the metabolomic analyses (Figure 2g,h). Supporting these results, an increase in ArgL activity was observed in a zebrafish PD model based on DJ-1 deficiency [66]. Changes in arg expression levels were also reported to have implications in the brain, although its role in this tissue is not yet known [67]. In addition, an enhancement of UC could serve to remove the excess of ammonia caused by the increased amino acid catabolism observed in DJ-1β mutant flies, as previously reported in Alzheimer’s disease [67].
This entry is adapted from the peer-reviewed paper 10.3390/cells11030331
References
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303.
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397.
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013.
- Panicker, N.; Ge, P.; Dawson, V.L.; Dawson, T.M. The cell biology of Parkinson’s disease. J. Cell Biol. 2021, 220, e202012095.
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28.
- Solana-Manrique, C.; Sanz, F.J.; Ripollés, E.; Bañó, M.C.; Torres, J.; Muñoz-Soriano, V.; Paricio, N. Enhanced activity of glycolytic enzymes in Drosophila and human cell models of Parkinson’s disease based on DJ-1 deficiency. Free Radic. Biol. Med. 2020, 158, 137–148.
- Anandhan, A.; Jacome, M.S.; Lei, S.; Hernandez-Franco, P.; Pappa, A.; Panayiotidis, M.I.; Powers, R.; Franco, R. Metabolic dysfunction in Parkinson’s disease: Bioenergetics, redox homeostasis and central carbon metabolism. Brain Res. Bull. 2017, 133, 12–30.
- Weissbach, A.; Wittke, C.; Kasten, M.; Klein, C. “Atypical” Parkinson’s disease-genetic. Int. Rev. Neurobiol. 2019, 149, 207–235.
- Niemann, N.; Jankovic, J. Juvenile parkinsonism: Differential diagnosis, genetics, and treatment. Parkinsonism Relat. Disord. 2019, 67, 74–89.
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259.
- Biosa, A.; Sanchez-Martinez, A.; Filograna, R.; Terriente-Felix, A.; Alam, S.M.; Beltramini, M.; Bubacco, L.; Bisaglia, M.; Whitworth, A.J. Superoxide dismutating molecules rescue the toxic effects of PINK1 and parkin loss. Hum. Mol. Genet. 2018, 27, 1618–1629.
- Van der Vlag, M.; Havekes, R.; Heckman, P.R.A. The contribution of Parkin, PINK1 and DJ-1 genes to selective neuronal degeneration in Parkinson’s disease. Eur. J. Neurosci. 2020, 52, 3256–3268.
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M.M. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 683920.
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s disease: Clinical insights and therapeutic perspectives. J. Clin. Med. 2019, 8.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Lotankar, S.; Prabhavalkar, K.S.; Bhatt, L.K. Biomarkers for Parkinson’s disease: Recent advancement. Neurosci. Bull. 2017, 33, 585–597.
- Havelund, J.F.; Heegaard, N.H.H.; Færgeman, N.J.K.; Gramsbergen, J.B. Biomarker research in Parkinson’s disease using metabolite profiling. Metabolites 2017, 7, 42.
- Binder, T.; Hobert, M.A.; Pfrommer, T.; Leks, E.; Granert, O.; Weigl, B.; Ethofer, T.; Erb, M.; Wilke, M.; Maetzler, W.; et al. Increased functional connectivity in a population at risk of developing Parkinson’s disease. Parkinsonism Relat. Disord. 2021, 92, 1–6.
- Li, X.; Fan, X.; Yang, H.; Liu, Y. Review of metabolomics-based biomarker research for Parkinson’s disease. Mol. Neurobiol. 2021, 1–17.
- Shukla, A.K.; Ratnasekhar, C.; Pragya, P.; Chaouhan, H.S.; Patel, D.K.; Chowdhuri, D.K.; Mudiam, M.K.R. Metabolomic analysis provides insights on paraquat-induced Parkinson-like symptoms in Drosophila melanogaster. Mol. Neurobiol. 2016, 53, 254–269.
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459.
- Bhinderwala, F.; Lei, S.; Woods, J.; Rose, J.; Marshall, D.D.; Riekeberg, E.; Leite, A.D.L.; Morton, M.; Dodds, E.D.; Franco, R.; et al. Metabolomics Analyses from Tissues in Parkinson’s Disease. Methods Mol. Biol. 2019, 1996, 217–257.
- Solana-Manrique, C.; Moltó, M.D.; Calap-Quintana, P.; Sanz, F.J.; Llorens, J.V.; Paricio, N. Drosophila as a model system for the identification of pharmacological therapies in neurodegenerative diseases. In Insights into Human Neurodegeneration: Lessons Learnt from Drosophila; Mutsuddi, M., Mukrherjee, A., Eds.; Springer Nature Pte Ltd.: Singapore, 2019; pp. 433–467.
- Aryal, B.; Lee, Y. Disease model organism for Parkinson disease: Drosophila melanogaster. BMB Rep. 2019, 52, 250–258.
- Lavara-Culebras, E.; Muñoz-Soriano, V.; Gómez-Pastor, R.; Matallana, E.; Paricio, N. Effects of pharmacological agents on the lifespan phenotype of Drosophila DJ-1beta mutants. Gene 2010, 462, 26–33.
- Lavara-Culebras, E.; Paricio, N. Drosophila DJ-1 mutants are sensitive to oxidative stress and show reduced lifespan and motor deficits. Gene 2007, 400, 158–165.
- Casani, S.; Gómez-Pastor, R.; Matallana, E.; Paricio, N. Antioxidant compound supplementation prevents oxidative damage in a Drosophila model of Parkinson’s disease. Free Radic. Biol. Med. 2013, 61, 151–160.
- Zhou, S.; Morgante, F.; Geisz, M.S.; Ma, J.; Anholt, R.R.H.; Mackay, T.F.C. Systems genetics of the Drosophila metabolome. Genome Res. 2020, 30, 392–405.
- Yon, M.; Decoville, M.; Sarou-Kanian, V.; Fayon, F.; Birman, S. Spatially-resolved metabolic profiling of living Drosophila in neurodegenerative conditions using 1H magic angle spinning NMR. Sci. Rep. 2020, 10, 9516.
- Yakhine-Diop, S.M.S.; Morales-García, J.A.; Niso-Santano, M.; González-Polo, R.A.; Uribe-Carretero, E.; Martinez-Chacon, G.; Durand, S.; Maiuri, M.C.; Aiastui, A.; Zulaica, M.; et al. Metabolic alterations in plasma from patients with familial and idiopathic Parkinson’s disease. Aging 2020, 12, 16690–16708.
- López Del Amo, V.; Palomino-Schätzlein, M.; Seco-Cervera, M.; García-Giménez, J.L.; Pallardó, F.V.; Pineda-Lucena, A.; Galindo, M.I. A Drosophila model of GDAP1 function reveals the involvement of insulin signalling in the mitochondria-dependent neuromuscular degeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 801–809.
- Bertrand, M.; Decoville, M.; Meudal, H.; Birman, S.; Landon, C. Metabolomic Nuclear Magnetic Resonance studies at presymptomatic and symptomatic stages of Huntington’s disease on a Drosophila model. J. Proteome Res. 2020, 19, 4034–4045.
- Figura, M.; Kuśmierska, K.; Bucior, E.; Szlufik, S.; Koziorowski, D.; Jamrozik, Z.; Janik, P. Serum amino acid profile in patients with Parkinson’s disease. PLoS ONE 2018, 13, e0191670.
- Andersen, A.D.; Binzer, M.; Stenager, E.; Gramsbergen, J.B. Cerebrospinal fluid biomarkers for Parkinson’s disease—A systematic review. Acta Neurol. Scand. 2017, 135, 34–56.
- Kumari, S.; Kumaran, S.S.; Goyal, V.; Sharma, R.K.; Sinha, N.; Dwivedi, S.N.; Srivastava, A.K.; Jagannathan, N.R. Identification of potential urine biomarkers in idiopathic parkinson’s disease using NMR. Clin. Chim. Acta 2020, 510, 442–449.
- Kumari, S.; Kumaran, S.S.; Goyal, V.; Bose, S.; Jain, S.; Dwivedi, S.N.; Srivastava, A.K.; Jagannathan, N.R. Metabolomic analysis of serum using proton NMR in 6-OHDA experimental PD model and patients with PD. Neurochem. Int. 2020, 134, 104670.
- LeWitt, P. Recent advances in CSF biomarkers for Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, S49–S51.
- LeWitt, P.A.; Li, J.; Lu, M.; Guo, L.; Auinger, P. Parkinson Study Group—DATATOP Investigators Metabolomic biomarkers as strong correlates of Parkinson disease progression. Neurology 2017, 88, 862–869.
- Trezzi, J.-P.; Galozzi, S.; Jaeger, C.; Barkovits, K.; Brockmann, K.; Maetzler, W.; Berg, D.; Marcus, K.; Betsou, F.; Hiller, K.; et al. Distinct metabolomic signature in cerebrospinal fluid in early parkinson’s disease. Mov. Disord. 2017, 32, 1401–1408.
- Tufi, R.; Gandhi, S.; de Castro, I.P.; Lehmann, S.; Angelova, P.R.; Dinsdale, D.; Deas, E.; Plun-Favreau, H.; Nicotera, P.; Abramov, A.Y.; et al. Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson’s disease. Nat. Cell Biol. 2014, 16, 157–166.
- Murali Mahadevan, H.; Hashemiaghdam, A.; Ashrafi, G.; Harbauer, A.B. Mitochondria in neuronal health: From energy metabolism to Parkinson’s disease. Adv. Biol. 2021, 5, e2100663.
- Garabadu, D.; Agrawal, N.; Sharma, A.; Sharma, S. Mitochondrial metabolism: A common link between neuroinflammation and neurodegeneration. Behav. Pharmacol. 2019, 30, 642–652.
- Shao, Y.; Le, W. Recent advances and perspectives of metabolomics-based investigations in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 3.
- Neinast, M.; Murashige, D.; Arany, Z. Branched chain amino acids. Annu. Rev. Physiol. 2019, 81, 139–164.
- Sinclair, E.; Trivedi, D.K.; Sarkar, D.; Walton-Doyle, C.; Milne, J.; Kunath, T.; Rijs, A.M.; de Bie, R.M.A.; Goodacre, R.; Silverdale, M.; et al. Metabolomics of sebum reveals lipid dysregulation in Parkinson’s disease. Nat. Commun. 2021, 12, 1592.
- Tosukhowong, P.; Boonla, C.; Dissayabutra, T.; Kaewwilai, L.; Muensri, S.; Chotipanich, C.; Joutsa, J.; Rinne, J.; Bhidayasiri, R. Biochemical and clinical effects of Whey protein supplementation in Parkinson’s disease: A pilot study. J. Neurol. Sci. 2016, 367, 162–170.
- Barichella, M.; Cereda, E.; Pinelli, G.; Iorio, L.; Caroli, D.; Masiero, I.; Ferri, V.; Cassani, E.; Bolliri, C.; Caronni, S.; et al. Muscle-targeted nutritional support for rehabilitation in patients with parkinsonian syndrome. Neurology 2019, 93, e485–e496.
- Szabó, N.; Kincses, Z.T.; Toldi, J.; Vécsei, L. Altered tryptophan metabolism in Parkinson’s disease: A possible novel therapeutic approach. J. Neurol. Sci. 2011, 310, 256–260.
- Gonzalez-Riano, C.; Saiz, J.; Barbas, C.; Bergareche, A.; Huerta, J.M.; Ardanaz, E.; Konjevod, M.; Mondragon, E.; Erro, M.E.; Chirlaque, M.D.; et al. Prognostic biomarkers of Parkinson’s disease in the Spanish EPIC cohort: A multiplatform metabolomics approach. NPJ Park. Dis. 2021, 7, 73.
- Ullah, R.; Jo, M.H.; Riaz, M.; Alam, S.I.; Saeed, K.; Ali, W.; Rehman, I.U.; Ikram, M.; Kim, M.O. Glycine, the smallest amino acid, confers neuroprotection against D-galactose-induced neurodegeneration and memory impairment by regulating c-Jun N-terminal kinase in the mouse brain. J. Neuroinflamm. 2020, 17, 303.
- García-Ayuso, D.; Di Pierdomenico, J.; Valiente-Soriano, F.J.; Martínez-Vacas, A.; Agudo-Barriuso, M.; Vidal-Sanz, M.; Picaud, S.; Villegas-Pérez, M.P. β-alanine supplementation induces taurine depletion and causes alterations of the retinal nerve fiber layer and axonal transport by retinal ganglion cells. Exp. Eye Res. 2019, 188, 107781.
- Kirov, I.I.; Sollberger, M.; Davitz, M.S.; Glodzik, L.; Soher, B.J.; Babb, J.S.; Monsch, A.U.; Gass, A.; Gonen, O. Global brain volume and N-acetyl-aspartate decline over seven decades of normal aging. Neurobiol. Aging 2021, 98, 42–51.
- Okarmus, J.; Havelund, J.F.; Ryding, M.; Schmidt, S.I.; Bogetofte, H.; Heon-Roberts, R.; Wade-Martins, R.; Cowley, S.A.; Ryan, B.J.; Færgeman, N.J.; et al. Identification of bioactive metabolites in human iPSC-derived dopaminergic neurons with PARK2 mutation: Altered mitochondrial and energy metabolism. Stem Cell Rep. 2021, 16, 1510–1526.
- Zilberter, Y.; Zilberter, M. The vicious circle of hypometabolism in neurodegenerative diseases: Ways and mechanisms of metabolic correction. J. Neurosci. Res. 2017, 95, 2217–2235.
- Hong, C.T.; Chau, K.-Y.; Schapira, A.H.V. Meclizine-induced enhanced glycolysis is neuroprotective in Parkinson disease cell models. Sci. Rep. 2016, 6, 25344.
- Díaz-García, C.M.; Yellen, G. Neurons rely on glucose rather than astrocytic lactate during stimulation. J. Neurosci. Res. 2019, 97, 883–889.
- Chakraborty, S.; Nian, F.-S.; Tsai, J.-W.; Karmenyan, A.; Chiou, A. Quantification of the metabolic state in cell-model of Parkinson’s disease by fluorescence lifetime imaging microscopy. Sci. Rep. 2016, 6, 19145.
- Messaoudi, N.; Gautier, V.; Dairou, J.; Mihoub, M.; Lelandais, G.; Bouloc, P.; Landoulsi, A.; Richarme, G. Fermentation and alternative respiration compensate for NADH dehydrogenase deficiency in a prokaryotic model of DJ-1-associated Parkinsonism. Microbiology 2015, 161, 2220–2231.
- Tapias, V.; McCoy, J.L.; Greenamyre, J.T. Phenothiazine normalizes the NADH/NAD+ ratio, maintains mitochondrial integrity and protects the nigrostriatal dopamine system in a chronic rotenone model of Parkinson’s disease. Redox Biol. 2019, 24, 101164.
- Cheng, Z.; Tsuda, M.; Kishita, Y.; Sato, Y.; Aigaki, T. Impaired energy metabolism in a Drosophila model of mitochondrial aconitase deficiency. Biochem. Biophys. Res. Commun. 2013, 433, 145–150.
- Cantu, D.; Schaack, J.; Patel, M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS ONE 2009, 4, e7095.
- Jodeiri Farshbaf, M.; Kiani-Esfahani, A. Succinate dehydrogenase: Prospect for neurodegenerative diseases. Mitochondrion 2018, 42, 77–83.
- Ikenaka, K.; Suzuki, M.; Mochizuki, H.; Nagai, Y. Lipids as trans-acting effectors for α-synuclein in the pathogenesis of Parkinson’s disease. Front. Neurosci. 2019, 13, 693.
- Simunovic, F.; Yi, M.; Wang, Y.; Macey, L.; Brown, L.T.; Krichevsky, A.M.; Andersen, S.L.; Stephens, R.M.; Benes, F.M.; Sonntag, K.C. Gene expression profiling of substantia nigra dopamine neurons: Further insights into Parkinson’s disease pathology. Brain J. Neurol. 2009, 132, 1795–1809.
- Pesi, R.; Balestri, F.; Ipata, P.L. Metabolic interaction between urea cycle and citric acid cycle shunt: A guided approach. Biochem. Mol. Biol. Educ. 2018, 46, 182–185.
- Edson, A.J.; Hushagen, H.A.; Frøyset, A.K.; Elda, I.; Khan, E.A.; Di Stefano, A.; Fladmark, K.E. Dysregulation in the brain protein profile of zebrafish lacking the Parkinson’s disease-related protein DJ-1. Mol. Neurobiol. 2019, 56, 8306–8322.
- Griffin, J.W.D.; Bradshaw, P.C. Amino acid catabolism in Alzheimer’s disease brain: Friend or foe? Oxid. Med. Cell. Longev. 2017, 2017, 5472792.
This entry is offline, you can click here to edit this entry!