Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Agriculture, Dairy & Animal Science
The COVID-19 virus strains were named after Greek alphabetical letters, and the designation is based on the positions and number of mutations. There are some disagreements regarding mutations belonging to specific strain groups, probably because different mutations evolved and spread further on different continents and states.
- biomarkers
- COVID-19
- epigenetics
- genetics
- miRNA
1. Virus Mutations
1.1. Virus Strains
Viral mutations and recombination gave birth to new strains that may have different reproduction rates or infectivity and may impact the course and severity of the disease.
The COVID-19 virus strains were named after Greek alphabetical letters, and the designation is based on the positions and number of mutations. There are some disagreements regarding mutations belonging to specific strain groups, probably because different mutations evolved and spread further on different continents and states. Mutations labeled with * are present in some strains [15]. The Alpha variant has mutations of sites E484K*, D614G, delH69V70, and N501Y. Next, the Beta strain, characterized by E484K, D614G, A701V, N501Y, L242_244L, and K417N mutations, and the Gamma strain, with mutations of sites E484K, D614G, K417T, N501Y, and T20, evolved, and both of them outcompeted the wild-type strain [16]. The Delta variant, with mutations of sites D614G, L452R, P681R, and T478K emerged in April 2021. In July 2021, the Delta variant outcompeted all the other strains [16]. Iota has mutations of sites A701V*, E484K*, L452R*, and D614G; Epsilon has mutations of sites L452R and D614G; Eta has mutations of sites E484K, D614G, and delH69V70; Kappa has mutations of sites E484Q, D614G, L452R, and P681R.
Another classification was made with slightly different mutations: Beta (B.1.351) has mutations of sites N501Y, E484K, K417N; Alpha (B.1.1.7) has mutations of sites N501Y, E484K, and HV69/70del; Gamma (P.1) has mutations of sites N501Y, E484K, K417T, and V1176F; Delta (B.1.617.2) has mutations of sites N501, E484, and (L452R, P681R, T478K) [17]. The third classification defines that the Delta strain holds L452R and T478K mutations, the Epsilon (B.1.427 and B.1.429) strain holds L452R mutations, and the Kappa (B.1.617.1) strain holds L452R and E484Q mutations [18].
1.2. Virus Mutations Position and Their Influence on SARS 2 Disease Development
SARS-CoV, which has a similar structure and RNA sequence to SARS-CoV2, had an estimated mutation rate of approximately 0.80–2.38 × 10–3 nucleotide substitutions per site per year, and the non-synonymous and synonymous substitution of approximately 1.16–3.30 × 10–3 and 1.67–4.67 × 10–3 per site per year, respectively, which is similar to other RNA viruses [20]. The large CoV RNA genome allows modification by introducing ‘’non-lethal’’ mutations and recombination, leading to increased probability for intraspecies variability, interspecies “host jump”, and novel CoVs to emerge [20]. SARS-CoV-2 has a higher fidelity in its transcription and replication process than other single-stranded RNA viruses because it has a proofreading mechanism, regulated by NSP14. However, despite this mechanism, the mutation rate is very high [21].
The mutation rate of SARS-CoV-2 is so high that it may impact diagnostic test accuracy [21]. In summary, the target spike and other SARS-CoV-2 proteins have numerous mutations. In total, 13,402 single mutations were found among 31,421 virus isolates, many of them located in coding regions currently used for COVID-19 diagnostic tests [21].
Out of 400 distinct mutation sites of spike protein, 10 mutation sites are most commonly mutated: D614(7859), L5(109), L54(105), P1263(61), P681(51), S477(57), T859(30), S221(28), V483(28), and A845(24) [22]. In Figure 2, mutations positions are presented on the 3D structure of activated mono-trimer spike protein (Figure 2A). Spike protein extracellular domain is divided into the S1 and S2 domains, on which RBD resides, and HR is divided into two domains, which connect S domains with the transmembrane region [23]. Mutations 483 and 477 are located on the loop close to the ACE2 binding site (Figure 2B). The rest are spread throughout the 3D structure of the spike protein, as shown in Figure 2C. Figures were prepared as described in the Supplementary Materials (Supplementary File S1).
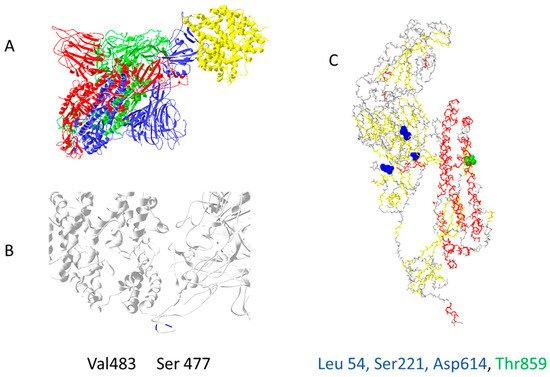
Figure 2. The most common mutation sites on activated spike protein: (A) trimer of activated spike protein (green, blue, red) interacts with ACE2 receptor (yellow); (B) two common mutation sites, V483 and S477 (dark blue), are close to the ACE2 interaction site; (C) three mutations should impact the 3D structure of spike proteins L54, S221, D614; T859 is located near spike trimer interaction site, PDB file—7DF4 [24].
Spike protein D614G mutation increases virus entry (Figure 3A) by enhancing the binding properties to the ACE2 protein [25]. Increased transmission is predicted for N501Y mutation [26] (Figure 3B). Residues 452, 489, 500, 501, and 505 on the RBM of spike protein (Figure 3C) have high chances of mutating into more infective strains [27]. Suleman et al. computed three mutations, N439K, S477 N, and T478K, shown in Figure 3D, to increase binding with ACE2 [28]. These mutations are localized on, or close to, the ACE2 binding site, except for D614G, which indirectly enhances ACE2 interaction through spike trimer binding enhancement (Figure 3). Bioinformatic predictions data match strain infectivity data. Mutations in strains overlap with the mutations predicted by bioinformatics tools [26,27,28].
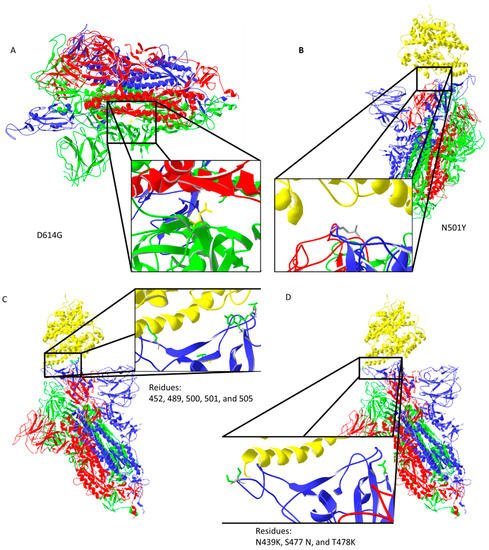
Figure 3. Important mutation sites of activated spike protein that increase or could increase the infectivity of the virus: (A) D614G mutation may increase interaction between trimer of spike protein (the negatively charged amino acid is replaced by neutral glycine that interacts with the opposite chain, where aliphatic amino acids are present); (B) N501Y mutation should reduce repulsive forces with ACE2; (C) mutations in the ACE2 interaction region could potentially increase the infectiveness of the virus; (D) predicted amino acid replacements that should increase binding with ACE2 are localized on the ACE2 binding site. Protein data bank (PDB) file: 7DF4 [24].
Information regarding 3D structure and mutagenesis is getting more accurate, which helps in drug development. Fast mutagenesis helps the virus evolve much faster than the human defense system can adapt to.
2. Virus–Host Interactions Affecting Viral Replication and Transcription
Viral infection triggers several mechanisms that are both virus- and host-dependent. On the one hand, viral replication affects transcription factors that promote viral replication; on the other hand, the host defense mechanism tries to activate factors to stop the virus from replicating itself.
SARS-CoV-2 targets transcription factors E2F1, SP1, EIF4A1, and TBP, and tumour suppressor genes, including PTEN, AKT1, and RB1, to influence the expression of different host genes (MAPK1, MAPK3, MAPK4, MAPK6, MAPK7, PIK3CA, and CAMK). For regulation, SARS-CoV-2 uses miRNAs [29]. Virus influence on RELA (NF-κB activation), E2F1, STAT3, TP53, NFKB1, GATA3, and CREB1 may impact the regulation of disease progression [30].
One CpG island with 18 CpG sites was detected at the start of viral RNA (UCSC Genome Browser). This site could impact the replication process and translation of proteins coded at the start of the RNA. If COVID-19 integrates into human DNA, this CpG island could impact when and how virus reactivation would start.
This entry is adapted from the peer-reviewed paper 10.3390/biology11020178
This entry is offline, you can click here to edit this entry!