Islet inflammation in T1DM is characterized by leukocyte infiltrates, in particular macrophages and T-cells which damage β-cells by release of cytokines, ROS and NO and also activation of death-receptor-mediated death pathways and subsequent phagocytosis. Production of cytokines such as INF-γ, TNFα and IL-1β act in synergy to promote elevation in concentration and increase in activity of NADPH oxidase and iNOS consequently increasing the formation of products including ROS and NO, respectively. The mechanism of action of INF-γ, TNFα and IL-1β involves stimulation of transcription factors including NFκB (in mouse islet β-cells).
- insulin
- metabolic reprogramming
- antidiabetic therapeutics
- glucose metabolism
- lipid metabolism
1. Introduction
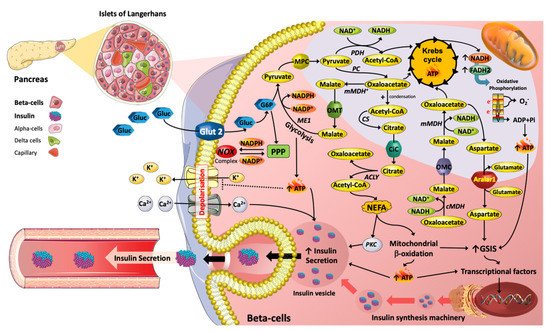
Pharmaceutical drug could induced metabolic adaptions/Reprogramming in β-Cells.
2. Antidiabetic Medications and Pancreatic β-Cell Metabolism
3. Pancreatic β-Cell G-Protein Coupled Receptors and Cell Metabolism
4. Contribution of Insulin, and Therapeutic Availability, to Metabolic Function of β-Cells
This entry is adapted from the peer-reviewed paper 10.3390/antiox11010108
References
- Marrano, N.; Biondi, G.; Cignarelli, A.; Perrini, S.; Laviola, L.; Giorgino, F.; Natalicchio, A. Functional loss of pancreatic islets in type 2 diabetes: How can we halt it? Metabolism 2020, 110, 154304.
- Yang, X.; Xu, Z.; Zhang, C.; Cai, Z.; Zhang, J. Metformin, beyond an insulin sensitizer, targeting heart and pancreatic β cells. Biochim. Biophys. Acta Mol. Bas. Dis. 2017, 1863, 1984–1990.
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270.
- El Messaoudi, S.; Rongen, G.A.; de Boer, R.A.; Riksen, N.P. The cardioprotective effects of metformin. Curr. Opin. Lipidol. 2011, 22, 445–453.
- Jiang, Y.; Huang, W.; Wang, J.; Xu, Z.; He, J.; Lin, X.; Zhou, Z.; Zhang, J. Metformin plays a dual role in MIN6 pancreatic β cell function through AMPK-dependent autophagy. Int. J. Biol. Sci. 2014, 10, 268–277.
- Leclerc, I.; Woltersdorf, W.W.; da Silva Xavier, G.; Rowe, R.L.; Cross, S.E.; Korbutt, G.S.; Rajotte, R.V.; Smith, R.; Rutter, G.A. Metformin, but not leptin, regulates AMP-activated protein kinase in pancreatic islets: Impact on glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E1023–E1031.
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investg. 2001, 108, 1167–1174.
- Hashemitabar, M.; Bahramzadeh, S.; Saremy, S.; Nejaddehbashi, F. Glucose plus metformin compared with glucose alone on β-cell function in mouse pancreatic islets. Biomed. Rep. 2015, 3, 721–725.
- Kitabchi, A.E.; Temprosa, M.; Knowler, W.C.; Kahn, S.E.; Fowler, S.E.; Haffner, S.M.; Andres, R.; Saudek, C.; Edelstein, S.L.; Arakaki, R.; et al. Role of insulin secretion and sensitivity in the evolution of type 2 diabetes in the diabetes prevention program: Effects of lifestyle intervention and metformin. Diabetes 2005, 54, 2404–2414.
- Lupi, R.; Del Guerra, S.; Fierabracci, V.; Marselli, L.; Novelli, M.; Patanè, G.; Boggi, U.; Mosca, F.; Piro, S.; Del Prato, S.; et al. Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes 2002, 51 (Suppl. S1), S134–S137.
- Lupi, R.; Del Guerra, S.; Tellini, C.; Giannarelli, R.; Coppelli, A.; Lorenzetti, M.; Carmellini, M.; Mosca, F.; Navalesi, R.; Marchetti, P. The biguanide compound metformin prevents desensitization of human pancreatic islets induced by high glucose. Eur. J. Pharmacol. 1999, 364, 205–209.
- Patanè, G.; Piro, S.; Rabuazzo, A.M.; Anello, M.; Vigneri, R.; Purrello, F. Metformin restores insulin secretion altered by chronic exposure to free fatty acids or high glucose: A direct metformin effect on pancreatic beta-cells. Diabetes 2000, 49, 735–740.
- Masini, M.; Anello, M.; Bugliani, M.; Marselli, L.; Filipponi, F.; Boggi, U.; Purrello, F.; Occhipinti, M.; Martino, L.; Marchetti, P.; et al. Prevention by metformin of alterations induced by chronic exposure to high glucose in human islet beta cells is associated with preserved ATP/ADP ratio. Diabetes Res. Clin. Pract. 2014, 104, 163–170.
- González-Barroso, M.M.; Anedda, A.; Gallardo-Vara, E.; Redondo-Horcajo, M.; Rodríguez-Sánchez, L.; Rial, E. Fatty acids revert the inhibition of respiration caused by the antidiabetic drug metformin to facilitate their mitochondrial β-oxidation. Biochim. Biophys. Acta 2012, 1817, 1768–1775.
- Lablanche, S.; Cottet-Rousselle, C.; Lamarche, F.; Benhamou, P.Y.; Halimi, S.; Leverve, X.; Fontaine, E. Protection of pancreatic INS-1 β-cells from glucose- and fructose-induced cell death by inhibiting mitochondrial permeability transition with cyclosporin A or metformin. Cell Deat. Dis. 2011, 2, e134.
- Jung, T.W.; Lee, M.W.; Lee, Y.J.; Kim, S.M. Metformin prevents endoplasmic reticulum stress-induced apoptosis through AMPK-PI3K-c-Jun NH2 pathway. Biochem. Biophys. Res. Commun. 2012, 417, 147–152.
- Simon-Szabó, L.; Kokas, M.; Mandl, J.; Kéri, G.; Csala, M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PLoS ONE 2014, 9, e97868.
- Dai, Y.L.; Huang, S.L.; Leng, Y. AICAR and Metformin Exert AMPK-dependent Effects on INS-1E Pancreatic β-cell Apoptosis via Differential Downstream Mechanisms. Int. J. Biol. Sci. 2015, 11, 1272–1280.
- He, X.; Gao, F.; Hou, J.; Li, T.; Tan, J.; Wang, C.; Liu, X.; Wang, M.; Liu, H.; Chen, Y.; et al. Metformin inhibits MAPK signaling and rescues pancreatic Aquaporin 7 expression to induce insulin secretion in type 2 diabetes mellitus. J. Biol. Chem. 2021, 297, 101002.
- Moon, J.S.; Karunakaran, U.; Elumalai, S.; Lee, I.K.; Lee, H.W.; Kim, Y.W.; Won, K.C. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J. Diabetes Complicat. 2017, 31, 21–30.
- Modak, M.A.; Parab, P.B.; Ghaskadbi, S.S. Control of hyperglycemia significantly improves oxidative stress profile of pancreatic islets. Islets 2011, 3, 234–240.
- McKinnon, C.M.; Docherty, K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia 2001, 44, 1203–1214.
- Jara, M.A.; Werneck-De-Castro, J.P.; Lubaczeuski, C.; Johnson, J.D.; Bernal-Mizrachi, E. Pancreatic and duodenal homeobox-1 (PDX1) contributes to β-cell mass expansion and proliferation induced by Akt/PKB pathway. Islets 2020, 12, 32–40.
- Obafemi, T.O.; Jaiyesimi, K.F.; Olomola, A.A.; Olasehinde, O.R.; Olaoye, O.A.; Adewumi, F.D.; Afolabi, B.A.; Adewale, O.B.; Akintayo, C.O.; Ojo, O.A. Combined effect of metformin and gallic acid on inflammation, antioxidant status, endoplasmic reticulum (ER) stress and glucose metabolism in fructose-fed streptozotocin-induced diabetic rats. Toxicol. Rep. 2021, 8, 1419–1427.
- Pan, Q.R.; Li, W.H.; Wang, H.; Sun, Q.; Xiao, X.H.; Brock, B.; Schmitz, O. Glucose, metformin, and AICAR regulate the expression of G protein-coupled receptor members in INS-1 beta cell. Horm. Metab. Res. 2009, 41, 799–804.
- Cho, Y.M.; Kieffer, T.J. New aspects of an old drug: Metformin as a glucagon-like peptide 1 (GLP-1) enhancer and sensitiser. Diabetologia 2011, 54, 219–222.
- Maida, A.; Lamont, B.J.; Cao, X.; Drucker, D.J. Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-α in mice. Diabetologia 2011, 54, 339–349.
- Brown, J.B.; Conner, C.; Nichols, G.A. Secondary failure of metformin monotherapy in clinical practice. Diabetes Care 2010, 33, 501–506.
- Kahn, S.E.; Haffner, S.M.; Heise, M.A.; Herman, W.H.; Holman, R.R.; Jones, N.P.; Kravitz, B.G.; Lachin, J.M.; O’Neill, M.C.; Zinman, B.; et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med. 2006, 355, 2427–2443.
- Matthews, D.A.-O.; Del Prato, S.; Mohan, V.; Mathieu, C.; Vencio, S.; Chan, J.C.N.; Stumvoll, M.; Paldánius, P.M. Insights from VERIFY: Early Combination Therapy Provides Better Glycaemic Durability Than a Stepwise Approach in Newly Diagnosed Type 2 Diabetes. Diabetes Ther. 2020, 11, 2465–2476.
- Ball, A.J.; Flatt, P.R.; McClenaghan, N.H. Desensitization of sulphonylurea- and nutrient-induced insulin secretion following prolonged treatment with glibenclamide. Eur. J. Pharmacol. 2000, 408, 327–333.
- Maedler, K.; Carr, R.D.; Bosco, D.; Zuellig, R.A.; Berney, T.; Donath, M.Y. Sulfonylurea induced beta-cell apoptosis in cultured human islets. J. Clin. Endocrinol. Metab. 2005, 90, 501–506.
- Wei, R.; Cui, X.; Feng, J.; Gu, L.; Lang, S.; Wei, T.; Yang, J.; Liu, J.; Le, Y.; Wang, H.; et al. Dapagliflozin promotes beta cell regeneration by inducing pancreatic endocrine cell phenotype conversion in type 2 diabetic mice. Metabolism 2020, 111, 154324.
- Pugazhenthi, S.; Qin, L.; Bouchard, R. Dipeptidyl peptidase-4 inhibition in diabetic rats leads to activation of the transcription factor CREB in β-cells. Eur. J. Pharmacol. 2015, 755, 42–49.
- Delobel, M.; Dalle, S. G-protein–coupled receptors controlling pancreatic β-cell functional mass for the treatment of type 2 diabetes. Curr. Opin. Endocr. Metab. Res. 2021, 16, 113–118.
- Winzell, M.S.; Ahrén, B. G-protein-coupled receptors and islet function-implications for treatment of type 2 diabetes. Pharmacol. Ther. 2007, 116, 437–448.
- Persaud, S.J. Islet G-protein coupled receptors: Therapeutic potential for diabetes. Curr. Opin. Pharmacol. 2017, 37, 24–28.
- Zhu, L.; Rossi, M.; Cohen, A.; Pham, J.; Zheng, H.; Dattaroy, D.; Mukaibo, T.; Melvin, J.E.; Langel, J.L.; Hattar, S.; et al. Allosteric modulation of β-cell M(3) muscarinic acetylcholine receptors greatly improves glucose homeostasis in lean and obese mice. Proc. Natl. Acad. Sci. USA 2019, 116, 18684–18690.
- Usui, R.; Yabe, D.; Fauzi, M.; Goto, H.; Botagarova, A.; Tokumoto, S.; Tatsuoka, H.; Tahara, Y.; Kobayashi, S.; Manabe, T.; et al. GPR40 activation initiates store-operated Ca(2+) entry and potentiates insulin secretion via the IP3R1/STIM1/Orai1 pathway in pancreatic β-cells. Sci. Rep. 2019, 9, 15562.
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101.
- Tudurí, E.; Imbernon, M.; Hernández-Bautista, R.J.; Tojo, M.; Fernø, J.; Diéguez, C.; Nogueiras, R. GPR55: A new promising target for metabolism? J. Mol. Endocrinol. 2017, 58, R191–R202.
- McKillop, A.M.; Moran, B.M.; Abdel-Wahab, Y.H.; Flatt, P.R. Evaluation of the insulin releasing and antihyperglycaemic activities of GPR55 lipid agonists using clonal beta-cells, isolated pancreatic islets and mice. Br. J. Pharmacol. 2013, 170, 978–990.
- Romero-Zerbo, S.Y.; Rafacho, A.; Diaz-Arteaga, A.; Suarez, J.; Quesada, I.; Imbernon, M.; Ross, R.A.; Dieguez, C.; Fonseca, F.R.d.; Nogueiras, R. A role for the putative cannabinoid receptor GPR55 in the islets of Langerhans. J. Endocrinol. 2011, 211, 177–185.
- Liu, B.; Song, S.; Ruz-Maldonado, I.; Pingitore, A.; Huang, G.C.; Baker, D.; Jones, P.M.; Persaud, S.J. GPR55-dependent stimulation of insulin secretion from isolated mouse and human islets of Langerhans. Diabetes Obes. Metab. 2016, 18, 1263–1273.
- Moran, B.M.; Flatt, P.R.; McKillop, A.M. G protein-coupled receptors: Signalling and regulation by lipid agonists for improved glucose homoeostasis. Acta Diabetol. 2016, 53, 177–188.
- McCloskey, A.G.; Miskelly, M.G.; Moore, C.B.T.; Nesbit, M.A.; Christie, K.A.; Owolabi, A.I.; Flatt, P.R.; McKillop, A.M. CRISPR/Cas9 gene editing demonstrates metabolic importance of GPR55 in the modulation of GIP release and pancreatic beta cell function. Peptides 2020, 125, 170251.
- Simcocks, A.C.; O’Keefe, L.; Jenkin, K.A.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. A potential role for GPR55 in the regulation of energy homeostasis. Drug Discov. Today 2014, 19, 1145–1151.
- Vong, C.T.; Tseng, H.H.L.; Kwan, Y.W.; Lee, S.M.; Hoi, M.P.M. G-protein coupled receptor 55 agonists increase insulin secretion through inositol trisphosphate-mediated calcium release in pancreatic β-cells. Eur. J. Pharmacol. 2019, 854, 372–379.
- Leibiger, I.B.; Leibiger, B.; Berggren, P.O. Insulin signaling in the pancreatic beta-cell. Annu. Rev. Nutr. 2008, 28, 233–251.
- Aspinwall, C.A.; Lakey, J.R.; Kennedy, R.T. Insulin-stimulated insulin secretion in single pancreatic beta cells. J. Biol. Chem. 1999, 274, 6360–6365.
- Johnson, J.D.; Misler, S. Nicotinic acid-adenine dinucleotide phosphate-sensitive calcium stores initiate insulin signaling in human beta cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14566–14571.
- Luciani, D.S.; Johnson, J.D. Acute effects of insulin on beta-cells from transplantable human islets. Mol. Cell Endocrinol. 2005, 241, 88–98.
- Khan, F.A.; Goforth, P.B.; Zhang, M.; Satin, L.S. Insulin activates ATP-sensitive K(+) channels in pancreatic beta-cells through a phosphatidylinositol 3-kinase-dependent pathway. Diabetes 2001, 50, 2192–2198.
- Hagren, O.I.; Tengholm, A. Glucose and insulin synergistically activate phosphatidylinositol 3-kinase to trigger oscillations of phosphatidylinositol 3,4,5-trisphosphate in beta-cells. J. Biol. Chem. 2006, 281, 39121–39127.
- Jonas, J.C.; Plant, T.D.; Gilon, P.; Detimary, P.; Nenquin, M.; Henquin, J.C. Multiple effects and stimulation of insulin secretion by the tyrosine kinase inhibitor genistein in normal mouse islets. Br. J. Pharmacol. 1995, 114, 872–880.
- Zawalich, W.S.; Zawalich, K.C. Effects of glucose, exogenous insulin, and carbachol on C-peptide and insulin secretion from isolated perifused rat islets. J. Biol. Chem. 2002, 277, 26233–26237.