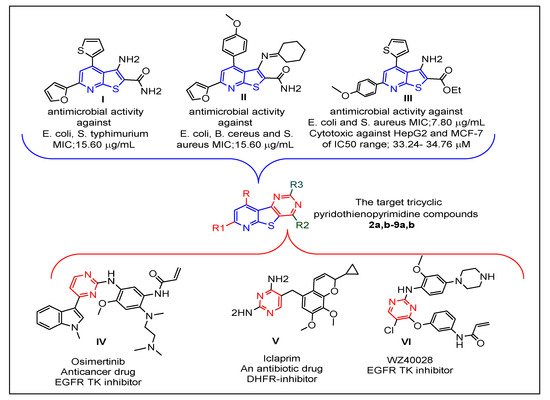
1. Introduction
In the past twenty years, the incidence of microbial infections associated with increased antimicrobial resistance has increased by alarming levels worldwide, endangering the possibility of curing many infectious diseases, and representing a global threat to population health [
1,
2,
3]. One of the potential approaches to combat this resistance problem is based on designing innovative new molecules with different modes of action to overcome cross-resistance to present therapeutics [
4,
5]. Efforts in developing broad-spectrum as well as specific and targeted drugs against various microbes are continuous in all directions [
5]. One of the recent strategies used for developing new antimicrobial agents is hybridization of different pharmacophores binding diverse biomolecular targets to exert synergistic effects against drug-resistant infectious diseases [
6].
On the other hand, despite large advances in the diagnosis and treatment of various types of cancers, the survival of cancer patients remains poor because of the widespread side effects of anticancer therapeutics as well as the acquisition of multiple drug resistance by the cancer cells [
7]. Breast and liver cancers are among the most medically significant cancers due to their high incidence and morbidity [
8]. Therefore, obtaining new, effective, more selective, and less toxic anticancer agents remains one of the most urgent demands [
9]. In addition, cancer patients are at higher risk of drug-resistant infectious diseases because of the weakness and immunosuppression caused by anticancer drug therapy [
10], which necessitates the development of new drugs that have potent dual activity against cancer and pathogenic microbes [
11].
Heterocyclic ring systems are considered key molecular structures in medicinal chemistry. In addition to their presence in the skeleton of many biological molecules such as hemoglobin, DNA, RNA, vitamins, hormones, and heterocyclic compounds [
12,
13,
14], they produce a wide range of biological activities [
15,
16]. Accordingly, the structural diversity and biological prominence of heterocyclic compounds have made them attractive synthetic targets in drug design and discovery for many years [
12,
13]. Thieno[2,3-
b]pyridines constitute a set of heterocyclic compounds that have beneficial effects in the treatment of many diseases. Recently, many studies have described various substituted thienopyridine compounds as significant antiproliferative agents against a wide range of human cancer cell lines [
17,
18,
19] and as antimicrobial [
20,
21,
22], anti-Alzheimer’s [
23], anti-platelet [
24], antiviral [
25,
26] and anti-inflammatory [
27] candidates.
Furthermore, the pyrimidine structure is closely related to the nucleobases—uracil, thymine, and cytosine—which makes pyrimidine molecules important building blocks in all living cells [
28,
29]. Pyrimidine-based compounds exhibit a broad spectrum of pharmacological activity, such as antimalarial [
30], antidiabetic [
31], antimicrobial [
32,
33], and anticancer activities [
34,
35,
36]. Therefore, combining thienopyridine and pyrimidine cores in the same molecular architecture, forming a pyridothienopyrimidine nucleus, serves as an attractive strategy for designing a novel scaffold with more favorable pharmacological effects [
37]. Recently, various pyridothienopyrimidine derivatives were reported to produce significant antimicrobial [
38,
39] and anticancer activities [
40,
41], as well as to suppress protein kinases such as serine/threonine kinase and vascular endothelial growth factor receptor (VEGFR-2) [
42,
43].
Prompted by the abovementioned issues, the strategy of this work was focused on the design of new tricyclic pyrido[3′,2′:4,5]thieno[3,2-
d]pyrimidine compounds with structures that hybridize features of the reported thienopyridine and pyrimidine antimicrobial and/or anticancer agents [
21,
22,
33,
34]
Figure 1. The resulting new pyridothienopyrimidines
2a,b–9a,b were synthesized via different cyclization reactions of the key starting compounds, 3-aminothieno[2,3-
b]pyridine-2-carboxamides
1a,b. The tricyclic ring system of the target pyridothienopyridine compounds was linked at position-2 and/or position-4 to different privileged structural motifs such as morpholine, piprazine, spiro cycloalkane, oxirane, and acetamide, which are renowned for their valuable and diverse pharmacological activity [
44,
45,
46,
47,
48]. All synthesized derivatives were evaluated as antimicrobial agents against a panel of Gram-positive and Gram-negative bacterial and fungal pathogens. Then, they were evaluated as cytotoxic agents against human liver carcinoma cells (HepG2) and human breast cancer cells (MCF-7) in an effort to gain new compounds possessing dual antimicrobial and anticancer activities. Since epidermal growth factor receptor (EGFR) is a key mediator in the regulation of different important cellular processes [
49,
50] and its overexpression displays a significant role in the development of many human cancers, including hepatic cancer and breast cancer [
51,
52], it was of interest to examine the EGFR inhibition activities of the compounds that revealed the most potent cytotoxic activities, as one of their cytotoxic mechanisms of action. Furthermore, molecular docking study was carried out to detect the compounds’ binding modes at the active site of EGFR-TK.
Figure 1. Reported thienopyridines (I–III) and pyrimidines (IV–VI) effective as antimicrobial and/or anticancer agents, and design of the new pyridothienopyrimidine compounds.
2. Insightful Analysis
2.1. Chemistry
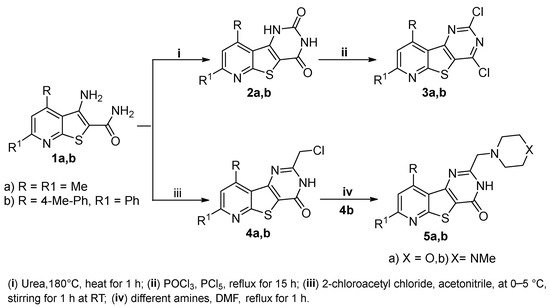
The synthetic pathways utilized for the synthesis of the target pyridothienopyrimidine compounds
2a,b–
9a,b are depicted in
Scheme 1 and
Scheme 2. The structures of all new compounds were confirmed using
1H-NMR,
13C-NMR (
Supplementary Materials), IR, and mass spectral data alongside the elemental microanalyses. Heating 3-amino-4,6-dimethyl/6-phenyl-4-(
p-tolyl)thieno[2,3-
b]pyridine-2-carboxamides
1a,b, the starting compounds, with urea at 180 °C for 1 h led to the formation of the corresponding pyridothienopyrimidine-2,4-diones
2a,b, which were further refluxed with a mixture of phosphorus oxychloride and phosphorus pentachloride to give the corresponding 2,4-dichloro derivatives
3a,b, respectively. On the other hand, the treatment of the starting compounds
1a,b with 2-chloroacetyl chloride in cold acetonitrile resulted in cyclization via substitution reaction followed by intramolecular elimination to give the 2-(chloromethyl)-pyridothienopyrimidin-4(3
H)-ones
4a,b, respectively. The subsequent treatment of
4b with different amines, namely, morpholine and 1-methylpiperazine, resulted in the corresponding 2-(morpholinomethyl)-/2-((4-methylpiperazin-1-yl)methyl)-pyridothieopyrimidin-4(3
H)-ones
5a,b (
Scheme 1). IR spectra of compounds
2a,b found two bands in the region 3394–3112 cm
−1 ascribed to 2NH and two bands in the region 1728–1640 cm
−1 related to the 2C=O groups of the pyrimidine-2,4(1
H,3
H)-dione ring. By comparison, IR spectra of the 2,4-dichloro derivatives
3a,b revealed the absence of the previous bands; instead, they showed two bands in the region 825–768 cm
−1 referring to the newly formed C-Cl groups. In addition,
1H-NMR spectra of compounds
2a,b revealed the two 2NH groups to be two D
2O exchangeable singlets at δ 10.42–11.71 ppm, which vanished in the
1H-NMR spectra of 2,4-dichloro derivatives
3a,b. Furthermore, the 2CH
3 of
2a,
3a and CH
3 of the p-tolyl moiety of
2b,
3b exhibited as singlet signals in the range 2.43–2.79 ppm along with aromatic protons in the range δ 7.21–8.24 ppm. The
13C NMR spectra of compounds
2a,b and
3a,b revealed CH
3 moieties at δ 20.40–24.36 ppm in addition to the aromatic carbons.
Scheme 1. Synthesis of pyridothienopyridine derivatives 2a,b–5a,b.
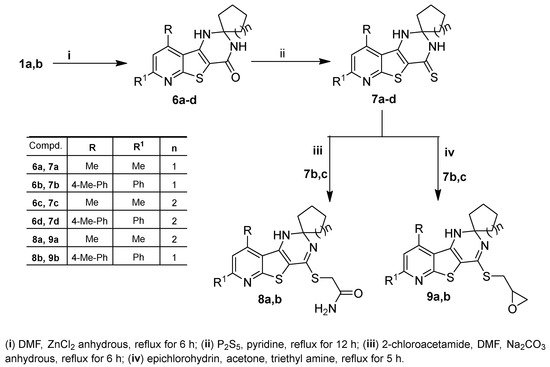
Scheme 2. Synthesis of 1′H-spirocycloalkane-1,2′-pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidine derivatives 6a–d–9a,b.
IR spectra of compounds 4a,b, 5a,b represented NH and C=O groups as absorption bands in the regions 3440–3387 cm−1 and 1669–1655 cm−1, respectively, while the C-Cl band of 4a appeared at 772 cm−1 and that of 4b was at 776 cm−1. Furthermore, 1H-NMR spectra of 4a,b and 5a,b exhibited, in addition to the signals of the parent protons, a singlet signal in the region δ 3.29–4.66, referring to the methylene protons of the -NCH2 moiety. Additionally, the eight protons of the morpholine moiety of 5a appeared as two singlets at δ 2.38 and 3.54 ppm, while those of the piperazine ring of 5b appeared as multiplet signals in the range δ 2.81–3.01 ppm alongside a singlet signal at δ 2.54 ppm due to its -N-CH3 group. The 13C-NMR spectra of 5a exhibited the 2CH2N- and 2CH2O of the morpholine moiety as two signals at δ 53.37 and 60.65 ppm, respectively.
The synthesis of spiro cycloalkane-pyridothienopyrimidin-4′-ones
6a–d in good yields was achieved via cyclocondensation reactions of the starting
1a,b with the appropriate cyclic ketones, cyclopentanone and/or cyclohexanone, in DMF containing zinc chloride anhydrous. Treating the latter compounds (
6a–d) with phosphorus pentasulfide in refluxing pyridine yielded the corresponding 1′
H-spiro[cycloalkane-1,2′-pyridothienopyrimidine]-4′(3′
H)-thiones
7a–d (
Scheme 2). IR spectra of
6a–d and
7a–d revealed the presence of different absorption bands in the regions 3427–3157 cm
−1 related to 2NH groups, 1660–1644 cm
−1 related to the C=O groups of
6a-d and 1247–1232 cm
−1 related to the C=S groups of compounds
7a–d.
1H-NMR spectra of
6a–d and
7a–d represented the eight protons of the spiro-cyclopentane and the ten protons of the spiro-cyclohexane substituents as multiplet signals in the range δ 1.21–2.11 ppm alongside one D
2O exchangeable singlet in the range δ 4.59–6.49 ppm, corresponding to the NH at position-1, and another D
2O exchangeable singlet appeared downfield in the range δ 7.81–9.85 ppm, related to the NH group at position-3.
13C-NMR spectra of
6a–d and
7a–d revealed various singlets in the ranges δ 19.26–38.33 ppm assignable to the cyclopentane and cyclohexane carbons, δ 69.49–70.81 ppm related to the spiro carbons, δ 164.40–167.80 ppm assigned to the C=O groups of
6a–d and δ 181.33–183.35 ppm ascribed to C=S moieties of compounds
7a–d, as well as the signals of the parent carbons, which appeared in their correct regions.
Compounds
7c,b were treated with 2-chloroacetamide in refluxing DMF containing sodium carbonate anhydrous to obtain the corresponding 4-sulfanyl acetamide derivatives
8a,b, while the treatment of
7c,b with epichlorohydrin in refluxing acetone containing a catalytic amount of triethyl amine resulted in the formation of the corresponding 4-(oxiran-2-ylmethy)sulfanyl derivatives
9a,b (
Scheme 2). The S-alkylation at position-4 was supported by
1H NMR and
13C-NMR spectra of
8a,b and
9a,b due to the vanishing of both the signal related to one of the two NH protons and that of the C=S carbon. Moreover, the
1H NMR spectra of compounds
8a,b exhibited singlet signals at δ 4.05 and 4.19 ppm due to SCH
2 protons of the newly formed acetamide side chain.
1H NMR spectra of compounds
9a,b represented the protons of the oxirane ring as two multiplets in the range δ 2.86–2.94 ppm, related to the methylene protons, and a third multiplet at δ 3.72–3.95 ppm, due to the methine proton, while the methylene protons of SCH
2 appeared as a doublet signal at δ 3.51 and 3.67 ppm. Additionally, the
13C-NMR spectra
9a,b showed three signals at the range δ 31.88–53.45 ppm referred to S
CH
2, O
CH
2, and O
CH moieties.
Confirmation of the molecular structures of the new compounds was also supported by their mass spectra, which represented the correct molecular ion peaks.
2.2. Biological Activity
2.2.1. Antimicrobial Activity
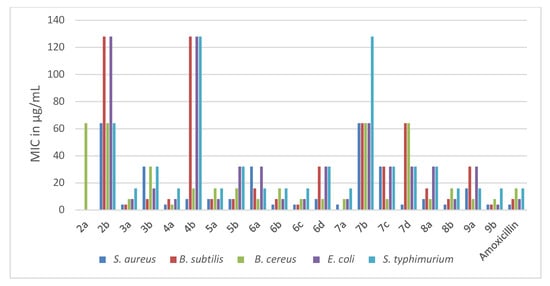
The newly synthesized pyridothienopyrimidine componds 2a,b–9a,b were investigated for their antimicrobial activities against a panel of microbial strains, three Gram-positive bacteria viz. Staphylococcus aureus 25923, Bacillus subtilis 6633, Bacillus cereus 33018, two Gram-negative bacteria viz. Escherichia coli 8739, Salmonella typhimurium 14028, three yeasts viz. Candida albicans 10231, Candida tropicals 750, Saccharomycese cerevisiae and two fungi viz. Aspergillus flavus, Aspergillus niger EM77 (KF774181). Their activities were expressed in terms of minimal inhibitory concentration (MIC) (μg/mL). Additionally, amoxicillin trihydrate and clotrimazole were utilized as positive antibiotic and antifungal controls. The obtained MIC results are represented in Table 1 and Table 2 and Figure 2 and Figure 3.
Figure 2. Antibacterial activities (MIC in µg/mL) of the new pyridothienpyrimidine compounds 2a,b–9a,b and the reference antibiotic (amoxicillin trihydrate).
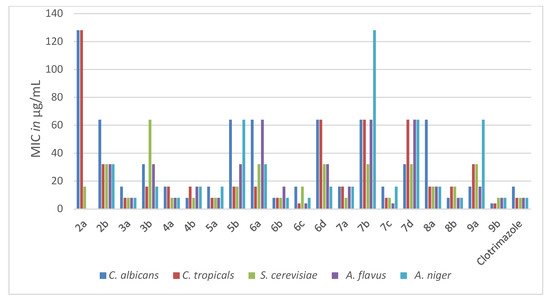
Figure 3. The antifungal activities (MIC in µg/mL) of the new pyridothienpyrimidine compounds 2a,b–9a,b and the reference drug (clotrimazole).
Table 1. In vitro antibacterial activities of the synthesized compounds expressed as MICs (μg/mL) against the tested pathogenic bacteria.
Table 2. In vitro antifungal activities of the synthesized compounds expressed as MICs (μg/mL) against the tested pathogenic yeasts and fungi.
Based on the MIC values in Table 1, the antibacterial activity of the target compounds 2a,b–9a,b was compared with that of amoxicillin, whose MICs ranged between 4–16 μg/mL against the five bacterial strains. It is evident that pyridothienopyrimidine-2,4(1H,3H)-dione derivatives 2a,b showed antibacterial activity varying from weak to inactive against the tested strains, with MICs ranging from 64 to >128 μg/mL. However, the conversion of 2a,b to 2,4-dichloro derivatives 3a,b led to a significant increase in antibacterial activity, especially for 7,9-dimethyl derivative 3a, which exhibited more potent activity than amoxicillin against B. subtilis and B. cereus and equalized with it against the other strains. In addition, 2-chloromethyl derivatives 4a,b revealed significant antibacterial activity, particularly 7,9-dimethyl derivative 4a, which showed the most potent activity against B. cereus with MIC = 4 μg/mL and had equipotent activity to amoxicillin against the other strains. While 7-Phenyl-9-(p-tolyl) analogue 4b revealed potent activity against S. aureus and B. cereus, it showed weak activity against other bacterial strains with MIC = 128 μg/mL. Further reaction of 2-chloromethyl derivative 4b with amines led to enhanced antibacterial activity, where 2-morpholinomethyl derivative 5a showed potent activity equal to amoxicillin against all the tested bacterial strains except S. aureus, with MICs ranging from 8 to 16 μg/mL. In addition, 2-(4-methylpiperazin-1-yl) derivative 5b revealed increasing activity against B. subtilis, E. coli and S. typhimurium.
Spiro cycloalkane-1,2′-pyridothienopyrimidin]-4′(3′H)-ones 6a–d exhibited antibacterial activity varying from potent to moderate against the tested strains, with MICs ranging from 4 to 32 μg/mL. Compound 6b showed an activity equal to that of the reference drug against the five bacterial strains, while 6c exceeded the potency of amoxicillin against B. subtilis and B. cereus and gave equipotent activity against the other strains. The other derivatives, 6a and 6d, showed activity varying from potent to moderate with MICs ranging 8–32 μg/mL. The conversion of 6a–d to 4′(3′H)-thiones analogues 7a–d resulted in an obvious weakening in the activity against the most of the tested strains, particularly derivative 7b, which showed weak activity against the five strains with MICs in the range 64–128 μg/mL. However, the S-alkylation of 7b and 7c at position-4 afforded increases in the antibacterial activity. The spiro cyclopentane 4-sulfanylacetamide derivative 8b revealed potent activity similar to that of amoxicillin, and the spiro cyclohexane analogue 8a showed potent to moderate activity with MICs ranging from 8 to 32 μg/mL. Spiro cyclopentane 4-(oxiran-2-ylmethyl)sulfanyl derivative 9b showed the most potent antibacterial activity against all tested strains, especially against E. coli, while spiro cyclohexane analogue 9a showed antibacterial activity ranging from potent to moderate with MICs in the range 8–32 μg/mL.
The antifungal activity of the new compounds 2a,b–9a,b was evaluated according to their MIC values in comparison to the MIC values of the reference antifungal drug clotrimazole, listed in Table 2. The target compounds (3a, 4a, 4b, 5a, 6b, 6c, 7a, 7c, 8b and 9b) appeared to be potent antifungal candidates, representing MIC values ranging from 4 to 16 μg/mL against all the tested yeasts and fungi strains, similar to the range of clotrimazole (MICs; 4–16 μg/mL). Furthermore, 4-(oxiran-2-ylmethyl)sulfanyl derivative 9b represented more potent antifungal activity than that of clotrimazole against the yeast pathogens C. albicans and C. tropicals, with MICs of 4 and 4 μg/mL, respectively. The rest of the target compounds displayed moderate to weak activity against the tested yeasts and fungi strains. The obtained results suggested that the conjugation of chloroine, chloromethyl, morpholinomethyl and spiro cyclopentane/cyclohexane at position-2 of the parent pyridothienopyrimid-4(3H)-one (3a, 4a,b, 5a, 6b,c, 7c) as well as the attachment of (oxiran-2-ylmethyl)sulfanyl and/or sulfanylacetamide side chains at position-4 of the pyridothienopyrimidine scaffold (8b, 9b) produced a beneficial impact for antimicrobial activity, resulting in promising broad-spectrum growth inhibition activity against the examined Gram-positive and Gram-negative microbes as well as fungal pathogens.
2.2.2. Cytotoxic Activity
The newly synthesized pyridothienopyrimidine derivatives
2a,b–
9a,b were subjected to antiproliferative activity evaluation against two cancer cell lines, hepatocellular carcinoma (HepG2) and breast cancer (MCF-7), by using the MTT colorimetric assay[
53]. The cytotoxic activity of the target compounds was compared with that of doxorubicin, utilized as a positive control. The concentrations of the examined derivatives that induced 50% inhibition of cell viability (IC
50, μM) were detected and are listed in
Table 3.
Table 3. Cytotoxic activities (IC50 µM) of the new compounds and doxorubicin against HepG2, MCF7 and WISH cells.
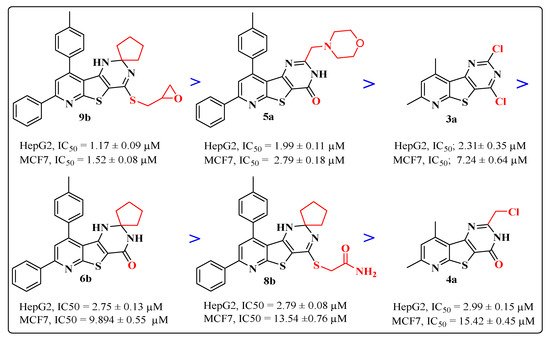
Based on IC50 values from Table 3, the examined compounds displayed versatile anti-proliferative activities against the tested cell lines, producing more potent growth inhibitory activity against HepG2 cells than MCF-7 cells with IC50 ranges of 1.17–56.18 µM and 1.52–77.41μM, respectively, compared to doxorubicin’s IC50 values of 2.85 and 3.58 µM, respectively. The most active cytotoxic activity was exhibited by the target compounds (9b, 5a, 3a, 6b, 8b and 4a), listed in descending order according to their activity against the two cell lines, as represented in Figure 4. Interestingly, the attachment of the sulfanylmethyl-oxirane side chain at position-4 of the spiro cyclopenane-1,2′-pyridothienopyrimidine nucleus in compound 9b produced the most potent growth inhibition activity against both HepG2 and MCF-7 cancer cells, approximately 2–3 fold higher than doxorubicin, representing IC50 values of 1.17 and 1.52 μM, respectively. However, the activity slightly decreased against HepG2 cells and detectably decreased against MCF-7 for the spiro cyclohexane sulfanylmethyl oxirane analogue 9a compared to doxorubicin, with IC50 values of 4.88 and 23.56 µM, respectively. Furthermore, the incorporation of the morpholine nucleus into the pyridothienopyrimidine scaffold at position-2 in compound 5a led to a significant cytotoxic potency against both HepG2 and MCF-7, greater than that obtained from the reference drug, at IC50 values of 1.99 and 2.79 µM, respectively. However, the 4-methylpiperazinyl analogue 5b exhibited an observable reduction in growth inhibition activity towards both tested cancer cell lines with IC50 values of 10.16 and 21.06 µM, respectively. A comparable growth inhibitory activity to doxorubicin was displayed against HepG2 cancer cells by the 2,4-dichloro-pyridothienopyrimidine derivative 3a at an IC50 value of 2.31 μM, but its activity was less against MCF-7 with an IC50 value of 7.24 µM, while its 7-phenyl-9-p-tolyl analogue 3b represented a detectable drop in potency, with IC50 values of 11.34 and 24.72 µM against HepG2 and MCF-7, respectively. The spirocyclopentane-pyridothienopyrimidin-4-one derivative 6b was nearly equipotent to doxorubicin in inhibiting the growth of HepG2 cancer cells, with an IC50 value of 2.75 µM, but produced a mild decrease in activity against MCF-7 with IC50 of 9.89 µM; however, the displacement of the spiro pentane moiety with spirocyclohexane in the analogue 6d led to a twofold decrease in cytotoxic activity against HepG2 cells and a significant drop in potency against MCF-7 cancer cells with IC50 values of 4.45 and 21.67 µM, respectively. The other members in this series, 6a,c, appeared to be significantly less potent candidates than the reference drug, with IC50 ranges of 12.11–77.41 µM.
Figure 4. The cytotoxic activity of the most potent compounds.
Additionally, the spiro cyclopentane sulfanylacetamide 8b produced an equivalent cytotoxic potency to that obtained by doxorubicin against HepG2 cells, with an IC50 value of 2.79 µM, but less potency than doxorubicin against MCF7, with an IC50 value of 13.54 µM. The spiro cyclohexane sulfanylacetamide analogue 8a appeared to be a less potent growth inhibitor towards both HepG2 and MCF-7 cancer cells, with IC50 values of 6.78 and 20.88 µM, respectively. Finally, the 2-chloromethyl-7,9-dimethyl derivative 4a, the last among the most potent compounds, showed significant growth inhibitory activity against HepG2 cancer cells with IC50 = 2.99 µM, but its potency decreased against MCF-7 cells with an IC50 value of 15.42 µM, whereas a high drop in activity was shown by the 7-phenyl-9-p-tolyl analogue 4b towards the two cancer cell lines with IC50 values of 36.52 and 43.27 µM, respectively. The rest of the target compounds, the spiro[cycloalkane-1,2′-pyridothienopyrimidine-4′(3′H)-thiones 7a–d and the pyridothienopyrimidine-2,4(1H,3H)-diones 2a,b, showed moderate to weak cytotoxic activities in the IC50 range of 10.35–41.62 µM, compared to doxorubicin.
The frequency and severity of undesirable effects on normal cells at therapeutic doses are considered among the most important characteristics that differentiate anticancer drugs from each other. Accordingly, the cytotoxic activity of the most active compounds (3a, 4a, 5a, 6b, 8b, 9b) was evaluated against the normal WISH cell line (Human amnion normal Liver cells) via MTT assay as IC50 values in μM, listed in Table 3. The obtained results revealed that the tested compounds had IC50 values in the range 394.98–460.23 µM, nearly equal to that obtained by the reference drug doxorubicin, IC50 doxorubicin 432.10 µM, which confirmed the high safety of the most potent compounds towards normal cells.
2.2.3. In Vitro EGFR Enzyme Inhibition Assay
The most potent cytotoxic compounds (
3a, 4a, 5a, 6b, 8b, 9b) were subjected to further investigations of their inhibiting profiles against EGFR, in order to study one of their proposed modes of action as potent cytotoxic agents [
54]. Accordingly, these derivatives were assessed as EGFR kinase inhibitors, using erlotinib as a reference drug as it is one of the most potent EGFR inhibitors [
55]. The obtained results were expressed as IC
50 values (nM) (
Table 4). Interestingly, the most cytotoxic derivatives, spiro cyclopentane-1,2′ pyridothienopyrimidine–oxirane
9b and pyridothienopyrimidine–morpholine
5a, appeared to be 3–2 times more potent than erlotinib, representing IC
50 values of 7.27, 9.66 and 27.01 nM, respectively. The 2-chloromethyl-pyridothienopyrimidin-4-one derivative
4a displayed a slight decrease in inhibition activity, but remained 1.5-fold more potent than erlotinib. An evident drop in EGFR inhibition activity was observed for the rest of the examined derivatives (
3a,
6b,
8b) representing an IC
50 range of 38.44–53.57 nM.
Table 4. In vitro enzymatic inhibitory activity against EGFR kinase.
2.3. Molecular Docking Studies
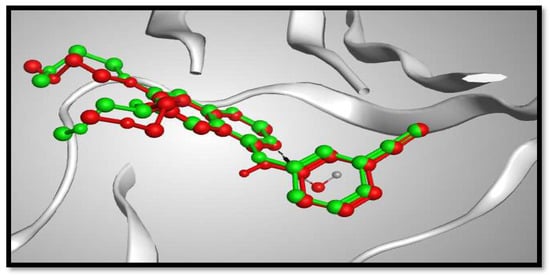
Molecular docking studies were performed to study the binding modes of the most potent cytotoxic compounds (3a, 4a, 5a, 6b, 8b, 9b) to the active sites of the target EGFR compared with erlotinib (ERL) as EGFR inhibitor. Docking setup was first validated through self-docking of the co-crystallized ligand (ERL) in the vicinity of the binding site of the enzyme. The docking score (S) was –10.48 kcal/mol. and root mean square deviation (RMSD) was 1.03 Å (Figure 5). The calculated RMSD value confirms the validity of the docking procedure.
Figure 5. 3D representation of the superimposition of the co-crystallized ligand (purple) and the docking pose (dark grey) of ERL at the active site of the EGFR enzyme.
Examination of the binding interactions of erlotinib to the active site of the EGFR kinase domain showed several conventional hydrogen bond interactions with the Met769, Leu768, Val702 and Leu820 amino acids (Figure 6).
Figure 6. (a) 2D interactions and (b) 3D interactions of erlotinib within the EGFR kinase domain’s active site.
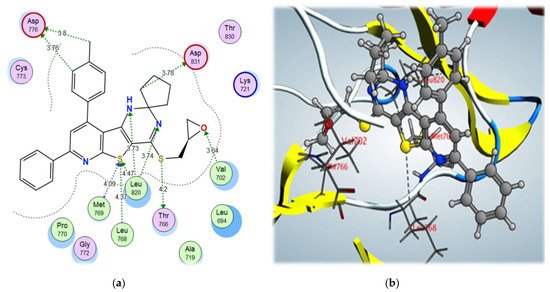
From the docking results of the examined compounds (Table 5), it was noticed that all compounds showed binding interactions within the active site of EGFR kinase domain with binding scores ranging from –12.01 to –8.94 kcal/mol. The spiro cyclopentane 4-((oxiran-2-ylmethyl)sulfanyl derivative 9b, which showed the highest biological activity, also showed the highest binding score of –12.01 kcal/mol, through binding with the key amino acids, Met769, Leu768, and Leu820, via the S atom of thiophene ring using a hydrogen bond acceptor, with further interactions with Thr766 via σ-hole bonding with the S atom of the S-methyl side chain. In addition, it bound to the amino acid Val702 through a hydrogen bond acceptor with the O atom of the oxirane moiety and showed high fitting in the vicinity of the active site, contributing to its high biological activity (Figure 7).
Figure 7. (a) 2D interactions and (b) 3D interactions of compound 9b within the EGFR kinase domain’s active site.
Table 5. Molecular docking results of the most active pyridothienopyrimidine compounds.
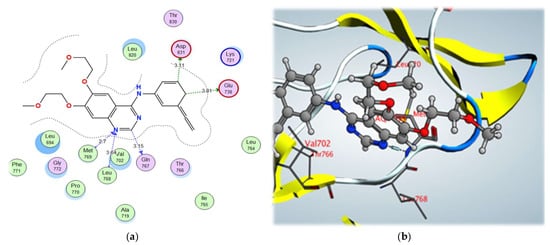
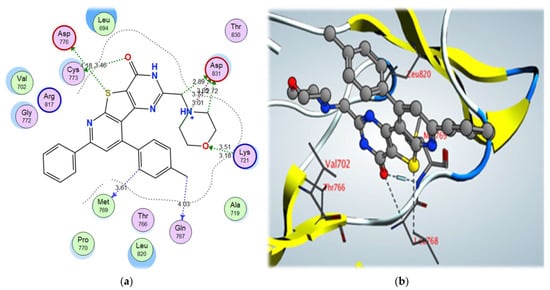
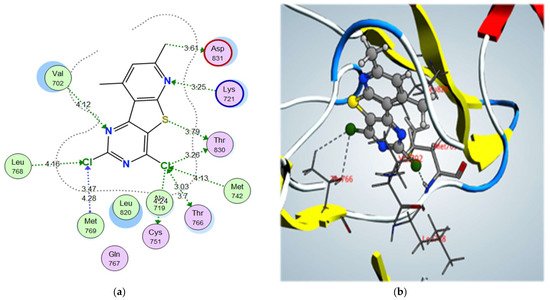
2-morpholinomethyl derivative 5a and 2,4-dichloro derivative 3a also showed higher binding scores than the co-crystallized ligand, erlotinib, at –11.48 and –11.42 kcal/mol, respectively; they showed a good binding pattern with the key amino acids, and compound 5a showed an ionic interaction with Asp831 revealing its high biological activity (Figure 8 and Figure 9). Other tested derivatives showed a lower binding score than erlotinib, with less binding interaction with the key amino acids.
Figure 8. (a) 2D interactions and (b) 3D interactions of compound 5a within the EGFR kinase domain’s active site.
Figure 9. (a) 2D interactions and (b) 3D interactions of compound 3a within the EGFR kinase domain’s active site.