Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Horticulture
Fusarium wilt disease is leading threat to watermelon yield and quality. Different cultivation cropping systems have been reported as safe and efficient methods to control watermelon Fusarium wilt. However, the role of salicylic acid (SA) in watermelon resistance to Fusarium wilt in these different cultivation systems remains unknown. In this experiment, we used RNA-seq and qRT-PCR to study the effect of SA biosynthesis on improving watermelon health, demonstrating how it may be responsible for Fusarium wilt resistance under continuous monocropping and oilseed rape rotation systems.
- salicylic acid
- watermelon
- Fusarium wilt
- resistance
1. Introduction
Watermelon (Citrullus lanatus) Fusarium wilt disease caused by Fusarium oxysporum f. sp. niveum (FON) poses a serious threat to watermelon quality and yield [1,2,3]. Symptoms such as rotted, discolored roots and brown vascular bundles appeared in Fusarium wilt diseased plants [4]. Different techniques include chemical control [5], biological control [6,7], grafting [8], and the use of disease-resistant cultivars [9,10] are utilized to overcome this disease. Many researchers found that different cultivation systems, such as intercropping [2,11], companion cropping [12], and rotated cropping [1], represent safe and efficient methods to control soil-borne diseases. Previously, researchers have focused on mutual recognition and recognition competition between the host plants, pathogens, pathogenic factors, and host plant defense factors. However, the molecular mechanisms involved therein remain unknown. Most researches have already identified the vital role of salicylic acid (SA) in plant defense [4,13] and programmed cell death [14]. For instance, reviews highlight the necessity of isochorismate synthase (ICS) [15] and phenylalanine ammonia-lyase (PAL) genes to synthesize SA and signal transduction mechanisms for activating the hypersensitive response and mediating system-acquired resistance (SAR) [13,16,17]. SA is also the main activator of defense response signal transduction [13,18]. Dou et al. determined that the expression of the genes involved in SA signal transduction increased, including the PR-1, WRKY, and TGA transcription factors, indicating that these genes play key roles in Foc4 infection [19]. Once certain levels of SA have been generated, SA-mediated activation of pathogen-related (PR) genes leads to a feedback in ICS expression in Arabidopsis, therefore preventing excessive SA accumulation [20,21]. Moreover, other studies show that the application with low level of exogenous SA could enhance plant resistance, while high concentration of it is harmful to plants [22,23]. Recently, Wang found that exogenous SA concentration >200 μM inhibited Foc TR4 growth [24]. Lv noticed that FON infection increased the accumulation of SA in watermelon [11]. Ren reported that exogenous SA (160 mg/L) could inhibit FON sporulation, but exogenously applied 100 μM of SA stimulate β-1,3-glucanase activity in watermelon leaves after FON inoculation [8]. Notably, our recent work revealed the important role of SA, JA, and ABA in resistance to watermelon Fusarium wilt. In addition, the results indicated that the SA content in the susceptible watermelon group was significantly higher than that in the resistant group [25].
2. Comparison of Disease Incidence, Watermelon Root Morphological Phenotype, and Physiological Indexes
In order to clarify the effect of exogenous SA on watermelon Fusarium disease resistance, the phenotypes of watermelon seedlings and the disease incidences after SA application at different concentrations at 7 dpi were explored. The observation of watermelon seedling results showed that there were severe plant yellowing and wilting symptoms in control group (0 μM, without SA treatment), while there were more leaves and developed roots after exogenous SA application at 100 μM as compared with it. However, a higher concentration of SA (200 μM) suppressed plant growth, shown by as the lower amount of leaves and roots of plants compared with 100 μM group (Figure 1A). In addition, the disease incidence in control group (0 μM, without SA treatment) was about 45%, but only 5% after exogenous SA application at 100 μM, which indicated the exogenous SA application could control the watermelon Fusarium disease occurrence (Figure 1B). Moreover, we compared the SA content after FON treatment with the control group in watermelon roots under the same growth conditions in the laboratory. The results showed that there was a significant increase in SA content in plants at 3 dpi and a descent after stabilization, but with a higher enrichment at the onset stage (7 dpi) (Figure 1C). Our data indicated that the SA was induced after FON attack in watermelon roots, suggesting that it may activate plant resistance at early stage. However, the reason for the SA accumulation at the onset stage might have been the disabled metabolisms or some modification activities [13].
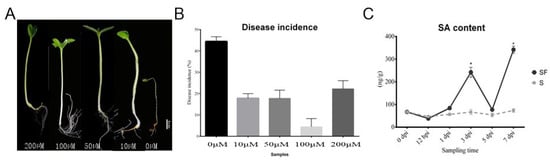
Figure 1. The effect of salicylic acid on watermelon resistance to Fusarium wilt in the pot experiment: (A) The phenotypes of watermelon seedling after SA application at different concentrations; (B) comparison of disease incidence after different concentrations of exogenous SA were applied; (C) comparison of SA content in watermelon treated with FON. S: control (Zaojia 8424, sensitive cultivar); SF: Zaojia 8424, sensitive cultivar + FON. Note: 0 dpi (before treatment); 12 hpi (12 h postinoculation); 1 dpi (1 day postinoculation); 3 dpi (3 days postinoculation); 5 dpi (5 days postinoculation); 7 dpi (7 days postinoculation). Data are expressed as mean ± SE (n = 3). Multiple t tests of two-way ANOVA (* p ≤ 0.0001).
Furthermore, the watermelon seedling morphology showed that the plants were much healthier and had more leaves and roots under rotation, as compared with those from the monocropping growth condition group (Figure 2A,B). Samples were referred to as C (continuous watermelon monocropping) and R (oilseed rape rotation cropping). The disease incidence results indicated that the incidence of watermelon Fusarium wilt in the rotated cropping system was significantly lower than that in the continuous cropping system group (Figure 2C). Moreover, the quantitative detection of FON in rhizosphere soil using real-time PCR [3,26] indicated that the abundance of FON exhibited no significant differences between these two systems (Figure 2D). This result suggested that there might be other reasons that led to plants being healthier in the rotated system, such as the structure of rhizosphere soil community [3,6,7]. The results from plant physiological indexes showed that the fresh root weight, root number, and root morphology were significantly affected. The fresh root weights (Figure 3A) in the R group were nearly twice those of the C group, and the pattern was the same for root number (Figure 3B). In addition, the results showed that there was a significant enhancement of the phenylalanine ammonia-lyase (PAL) enzyme activity (Figure 3D), but a decrease in peroxidase (POD) enzyme activity (Figure 3E) and malondialdehyde (MDA) content (Figure 3F) in the R group as compared with the C group. Therefore, these results showed that the plants under rotation (R) were healthier than those under the continuous cropping system (C). More importantly, our study showed a significantly higher SA content in the C group as compared to the R group, which confirmed that a higher level of SA was harmful to the plants (Figure 3C). Therefore, we aimed to further explore the different gene expression patterns related to SA pathways in watermelon roots that might be responsible for Fusarium wilt resistance under different cultivation systems.
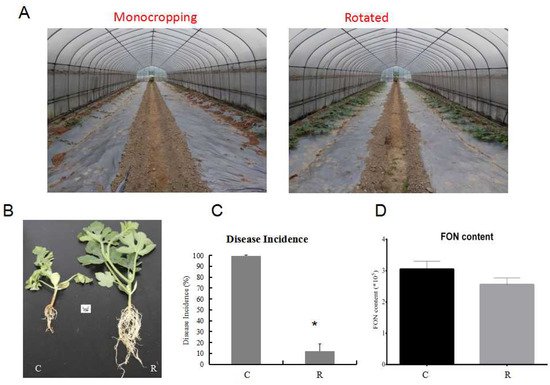
Figure 2. Comparison of watermelon growth and disease incidence under different growth conditions in the field experiment. (A) Comparison of watermelon plants’ morphology under monocropping and rotated cropping systems; (B) the phenotypes of watermelon seedlings under different systems; (C) the disease incidence under different growth systems; (D) FON biomass in two cropping systems. C: continuous watermelon monocropping; R: oilseed rape rotation cropping. Data are expressed as mean ± SE (n = 3). Student’s t-test (* p ≤ 0.05).
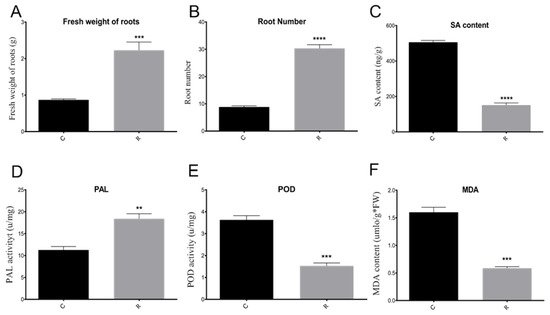
Figure 3. Comparison of watermelon physiological and biochemical indexes. (A) Comparison of fresh root weights in different samples; (B) comparison of root number in different samples; (C) comparison of SA contents in different samples; (D) comparison of PAL enzyme activity in different samples; (E) comparison of POD enzyme activity in different samples; (F) comparison of MDA content in different samples. C: continuous watermelon monocropping; R: rotated with oilseed rape cropping; POD: peroxidase; PAL: phenylalanine ammonia-lyase; MDA: malondialdehyde. Three biological replicates per samples were analyzed. Data are expressed as mean ± SE (n = 3). Student’s t-test (** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001).
3. Transcriptome Profiling of Watermelon Roots under Different Growth Conditions
We used transcriptome sequencing to analyze the molecular mechanisms of SA biosynthesis in watermelon plant roots activating Fusarium wilt resistance under different growth conditions. We randomly selected nine healthy plant roots using the five-spot sampling method from each greenhouse and mixed them as one replicate; we collected three replicates from three greenhouses. Samples were referred to as C (continuous watermelon monocropping) and R (oilseed rape rotation cropping). There were 21,910 genes detected and 5748 significantly deferentially expressed genes when comparing the C and R groups, with 3240 being upregulated and 2508 genes being downregulated (Figure 4A).
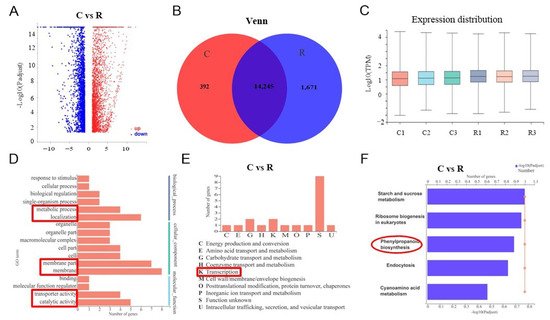
Figure 4. Transcriptome comparison analysis of watermelon gene expression under different cropping systems: (A) volcano map with different expressions; (B) Venn diagram of watermelon genes expression under different cropping systems; (C) box plot of gene expression distribution; (D) GO annotations analysis; (E) COG classification; (F) comparison analysis of major KEGG pathway enrichment. C: continuous watermelon monocropping; R: oilseed rape rotation cropping. Three biological replicates per samples were analyzed.
The Venn diagram and box plot show the list of expressed genes and their distribution under the two different systems (Figure 4B,C). The Venn diagram indicates that there were 14,245 genes were overlapped in C and R groups, and 392 significantly different expressed genes in C but 1671 significantly different expressed genes in R. The box plot shows that the gene expression distribution was uniform. The GO annotation analysis indicated that there were 17 significantly differentially expressed GO pathways compared under different groups in our experiment (Figure 4D). For instance, the metabolic process, localization, membrane, transporter activity, and catalytic activity (red markers in Figure 4D). The significantly expressed genes were merged into nine groups using COG classification, which mostly were in transcription (K) (Figure 4E). Further, the KEGG enrichment analysis revealed that five major pathways were significantly activated, including starch and sucrose metabolism, ribosome biogenesis in eukaryotes, endocytosis, cyanoamino acid metabolism, and especially phenylpropanoid biosynthesis (red marker in Figure 4F). Thus, our sequencing results supported the key role of SA in the resistance of watermelon to Fusarium wilt, indicating that it activated a series of defensive feedback mechanisms under environmental stress through several signal transduction pathways.
On the basis of the physiological indexes and gene function enrichment analysis, we analyzed the differential expression of various significantly expressed candidate genes related to pathogen resistance and SA biosynthesis. The results showed that there were increases in highly expressed genes in wound-induced proteins, peroxidases, and pathogenesis-related proteins in group C. However, genes related to stress-response proteins and other membrane-response proteins were highly expressed in group R (Figure 5A). Therefore, our results indicated that the continuous cropping environment may have induced the expression of certain peroxidases and pathogenesis-related genes, but the stress-response genes were not activated, which can lead to plant death. Moreover, our results illustrated that the main SA biosynthesis pathway in watermelon roots is the PAL pathway, and the upregulated expression of the CIPALs genes was an essential factor in SA accumulation in watermelon roots (Figure 5B).
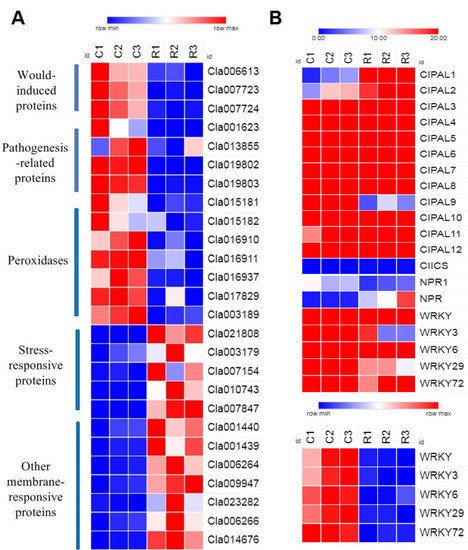
Figure 5. Differential expression genes in watermelon roots under different cropping systems: (A) comparison analysis of the expression of 26 genes under different cropping systems; (B) comparison analysis of the relative expression of 20 candidate genes in SA biosynthesis. The relative color scheme uses selected values in each row to convert values to colors. C: continuous watermelon monocropping; R: oilseed rape rotation cropping. Three biological replicates per samples were analyzed.
4. Expression Verification of SA Biosynthesis Genes
In order to further test the hypotheses concerning the different expressed genes related to SA biosynthesis in watermelon roots under different growth conditions, we examined the results of the RT-qPCR analysis. These data confirmed significant differences in the expressions of the CIPALs, NPR, and NPR1 genes (Figure 6). The results emphasized the important role of the CIPAL1, CIPAL9, and CIPAL12 genes in SA biosynthesis. NPR expression was significantly higher in group R, while NPR1 expression was higher in group C, suggesting that they may have different functions under different growth conditions.
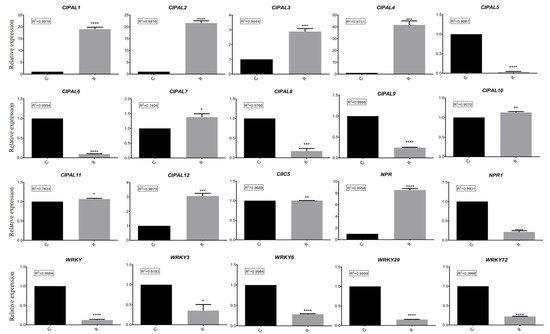
Figure 6. Comparison analysis of relative expressions of 20 candidate genes in different samples using RT-qPCR. C: continuous watermelon monocropping; R: oilseed rape rotation cropping. Three biological replicates per samples were analyzed. Data are expressed as mean ± SE (n = 3). Student’s t-test (* p ≤ 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001).
5. Bioinformatics Analysis of Candidate Genes
Three candidate SA synthesis genes were selected for protein homology analysis using NCBI BLAST. The results showed that the Cla008727 protein exhibited a high level of similarity with ABM 67591.1 | phenylalanine ammonia-lyase (Vitis vinifera L.) and XP_ 012082374.1 | phenylalanine ammonia-lyase (Jatropha curcas L.). The Cla013761 protein demonstrated a high level of similarity with Cucumis sativus L., Ricinus communis L., Populus trichocarpa (Torr. & Gray), Populus tomentosa Carrière, and Cucumis melo L. phenylalanine ammonia-lyase. In addition, the Cla 018303 protein had more than 90% similarity to many phenylalanine ammonia-lyase-like proteins in Cucumis sativus L. Therefore, our phylogenetic analysis showed that these candidate genes had the closest evolutionary relationship with Cucumis sativus L. and Cucumis melo L. (Figure 7). Six candidate genes related to SA metabolism were selected to analyze whether they possessed phytohormone cis-regulatory elements. Our results indicated that NPR and WRKY29 possessed an SA responsive element, whereas NPR1, WRKY, WRKY6, WRKY29, and WRKY72 possessed MeJA- and ABA-responsive elements (Table 1).
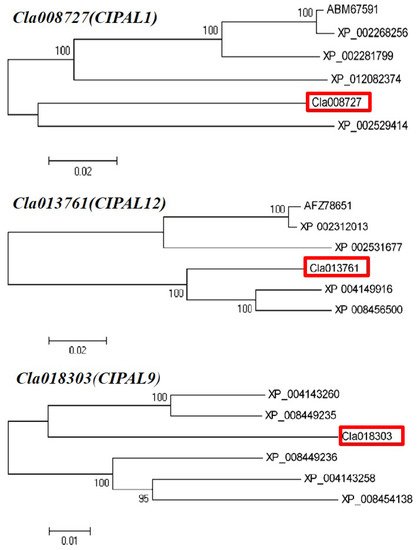
Figure 7. Phylogenic tree of the homologue’s proteins from three candidate genes.
Table 1. Prediction of cis-acting regulatory element of six DEGs.
| Gene ID | Gene Name | Cis-Acting Element |
Function | Sequence | Number |
|---|---|---|---|---|---|
| Cla002899 | NPR1 | ABRE | ABA | ACGTG | 1 |
| Cla019154 | NPR | CGTCA-motif | MeJA | CGTCA | 2 |
| TCA-element | SA | CCATCTTTTT, TCAGAAGAGG |
5 | ||
| Cla022362 | WRKY | ABRE | ABA | AACCCGG | 1 |
| CGTCA-motif | MeJA | CGTCA | 1 | ||
| Cla002084 | WRKY6 | CGTCA-motif | MeJA | CGTCA | 1 |
| Cla005515 | WRKY2 | ABRE | ABA | CACGTG, ACGTG | 5 |
| CGTCA-motif | MeJA | CGTCA | 2 | ||
| TCA-element | SA | CCATCTTTTT | 1 | ||
| Cla010867 | WRKY72 | ABRE | ABA | ACGTG | 2 |
| CGTCA-motif | MeJA | CGTCA | 3 |
This entry is adapted from the peer-reviewed paper 10.3390/plants11030293
This entry is offline, you can click here to edit this entry!