Peroxisomes are single membrane-bound organelles found in all eukaryotic cells and organisms, from yeast to plants and mammals. They are key regulators of cellular and metabolic homeostasis. These organelles play important roles in redox metabolism, the oxidation of very-long-chain fatty acids (VLCFAs), and the biosynthesis of ether phospholipids. Given the essential role of peroxisomes in cellular homeostasis, peroxisomal dysfunction has been linked to various pathological conditions, tissue functional decline, and aging. In the past few decades, a variety of cellular signaling and metabolic changes have been reported to be associated with defective peroxisomes, suggesting that many cellular processes and functions depend on peroxisomes. Peroxisomes communicate with other subcellular organelles, such as the nucleus, mitochondria, endoplasmic reticulum (ER), and lysosomes. These inter-organelle communications are highly linked to the key mechanisms by which cells surveil defective peroxisomes and mount adaptive responses to protect them from damages.
1. Introduction
Eukaryotic cells contain a variety of membrane-enclosed organelles. Among them, peroxisomes are ubiquitous single membrane-bound organelles that play essential roles in cellular metabolism. Peroxisomes are the major site for both the production and degradation of hydrogen peroxide (H
2O
2) due to their high content of peroxisomal oxidases and antioxidant enzymes, such as catalase. Peroxisomes are also critical for the biosynthesis of ether phospholipids, cholesterol, bile acids, and polyunsaturated fatty acids (PUFAs). The importance of peroxisomes is evidenced by peroxisomal biogenesis disorders (PBDs), rare genetic diseases caused by mutations in any of the 13 different
PEX genes encoding peroxins responsible for the import of membrane or matrix proteins to peroxisomes [
1]. Zellweger syndrome (ZS) is the most severe PBD, and patients with ZS show severe brain, liver, kidney, and bone malfunctions [
2].
Given the essential roles of peroxisomes in cellular and metabolic homeostasis [
3,
4], it is not surprising to find that peroxisomal dysfunction is strongly linked to the loss of redox homeostasis, dysregulated lipid metabolism, mitochondrial dysfunction, altered gene expression, and elevated ER stress and cell death. More recently, it has become clear that peroxisome deficiency can also alter functions of specific subcellular organelles [
5,
6,
7], such as mitochondria, endoplasmic reticulum (ER), and lysosome. Peroxisomal stress signals can also be transduced from peroxisomes to the nucleus, as peroxisomal dysfunction often induces global transcriptional changes [
8,
9]. Thus, similar to other organelle stress pathways (e.g., ER stress), there are specific peroxisomal stress response pathways that allow eukaryotic cells to cope with malfunctioning peroxisomes [
10].
Peroxisomes can communicate with other organelles through membrane contact sites, peroxisome-derived metabolites, as well as retrograde signaling. However, it remains largely unclear how peroxisomal stress response pathways are activated and how they protect cells from damages. Understanding peroxisomal stress response pathways and how peroxisomes communicate with other organelles are important areas of peroxisome research because of their vital role in cellular homeostasis, aging, and aging-related diseases [
11,
12,
13,
14].
2. Peroxisome Biogenesis and Peroxisomal Import Machinery
Peroxisomes are single membrane-bound organelles found in all eukaryotic cells and organisms, from yeast to plants and mammals [
15]. Under normal conditions, the peroxisome has a half-life of 1.5 to 2 days [
16], suggesting that peroxisome biogenesis and degradation are very dynamic processes. Peroxisomal biogenesis is mostly regulated by peroxins encoded by
PEX genes. To date, more than 30 conserved peroxins have been identified in yeast, plants, and many animal species [
17]. Peroxisomes can be formed by
de novo biogenesis from the ER and mitochondria [
17,
18,
19]. Pre-peroxisomal vesicles originating from the ER contain the peroxisomal membrane protein (PMP) PEX16, which binds to the PMP receptor PEX3. PEX3 functions as a docking receptor for the cytosolic chaperone and PMP receptor PEX19 [
20,
21]. Together, these peroxins recruit other PMPs containing membrane peroxisome targeting signals (PTS) to the peroxisomal membrane during the peroxisome maturation process.
Peroxisomal matrix proteins are transported into the organelle after synthesis in the cytosol [
22]. PTSs are essential for the correct sorting of matrix proteins [
23]. Most peroxisomal matrix proteins contain a PTS type 1 (PTS1) sequence at the C-terminus consisting of an S-K-L motif or a conservative variant [
24,
25]. A small set of matrix proteins contains a PTS type 2 (PTS2) sequence, a nonapeptide with the consensus sequence (R/K)-(L/I/V)-X
5-(H/Q)-(L/A) (X = any amino acid) that is localized near or at the
N-terminus of the protein [
26,
27]. PTS1 proteins are transported to the peroxisome by the shuttling receptor PEX5, whereas PTS2 proteins are delivered to the organelle by PEX5 and PEX7 [
23]. The cargo-loaded receptor proteins bind to the peroxisomal membrane proteins PEX13 and PEX14 [
28,
29]. After docking, the cargo proteins are translocated across the peroxisomal membrane and imported into the organelle lumen, while receptor protein PEX5 is then mono- or polyubiquitinated by E3 ubiquitin ligases PEX2, PEX10, and PEX12 [
23,
30,
31]. PEX5 monoubiquitination is required for its recycling to the cytosol, while polyubiquitinated PEX5 can be targeted to the proteasome for degradation [
32,
33,
34]. Cytosolic AAA-ATPases PEX1 and PEX6 form a heterohexameric ring as a trimer of PEX1/PEX6 heterodimers, which is anchored to the peroxisome membrane by PEX26 in higher organisms [
35,
36,
37,
38,
39]. The PEX1-PEX6-PEX26 complex is responsible for releasing ubiquitinated PEX5 from the peroxisomal membrane to the cytosol [
33,
34,
35]. In the cytosol, PEX5 can be deubiquitinated by deubiquitylating (DUB) enzymes (Ubp15 in yeast [
40] and USP9X in mammals [
41]) for the next cycle of peroxisomal import.
Mature peroxisomes divide and grow by elongation, membrane constriction, and fission [
22,
42,
43,
44,
45]. In mammals, the peroxisomal membrane protein PEX11β plays a critical role in peroxisomal proliferation [
42,
46,
47]. Exogenous expression of
PEX11β in mammalian cells enhances peroxisomal proliferation [
42], whereas the genetic defects of human
PEX11β decrease peroxisomal abundance [
47]. Although the molecular mechanisms underlying the modulation of PEX11β activity are not fully understood, docosahexaenoic acid (DHA) is known to play an important role in the initiation of peroxisomal elongation and fission [
48]. Itoyama et al. [
48] demonstrated that DHA, a peroxisomal β-oxidation metabolite, induced peroxisomal division by augmenting the hyper-oligomerization of Pex11pβ and the formation of Pex11pβ-enriched regions on elongated peroxisomes. The function of Pex11β in peroxisome proliferation depends on the homo-oligomerization through its
N-terminal domain [
46,
49]. In addition, Pex11β interacts with the membrane via the
N-terminal amphipathic helix to deform the membrane into elongated tubular peroxisomes [
50,
51,
52]. Subsequently, Pex11β forms a ternary fission machinery complex with mitochondrial fission factor (Mff) and dynamin-like protein 1 (DLP1) at the constricted membrane region of elongated peroxisomes, promoting the scission of the membrane [
51,
53].
3. Key Metabolic Functions of Peroxisome
Peroxisomes are multifunctional organelles that play essential roles in several key metabolic pathways, including the β-oxidation of very-long-chain fatty acids (VLCFAs, >C22), the α-oxidation of branched-chain fatty acids, the oxidation of D-amino acids and polyamines, and the synthesis of ether phospholipids, bile acids, and DHA [
2,
17]. During these metabolic processes (e.g., VLCFA β-oxidation), reactive oxygen species (ROS) are produced as byproducts and subsequently neutralized by several antioxidant enzymes such as catalase [
54,
55,
56]. About 50 different enzymes are involved in peroxisomal metabolic functions [
2,
57], and some of them are shared with other subcellular compartments, including mitochondria [
57]. Accordingly, cooperation with other organelles, including mitochondria, ER, lipid droplets, and lysosomes, is necessary for proper peroxisomal metabolic functions.
Most fatty acids (FAs) are catabolized by β-oxidation, which removes two carbons from their carboxyl terminus [
58]. In all eukaryotic cells, fatty acid oxidation occurs in both mitochondria and peroxisomes. FAs (<C20) are oxidized in mitochondria and converted to CO
2 and H
2O through the mitochondrial citric acid cycle and oxidative phosphorylation. However, VLCFAs cannot be oxidized in mitochondria. Instead, VLCFAs translocate across the peroxisomal membrane via ATP-binding cassette (ABC) subfamily D half-transporters (ABCD) and are broken down to acetyl-CoA, propionyl-CoA, or medium-chain acyl-CoAs. These different acyl-CoAs can be further converted into acylcarnitines in the peroxisome and translocate to mitochondria through the mitochondrial carnitine/acylcarnitine translocase (CACT). Acylcarnitines are then converted back to acyl-CoAs in mitochondria and enter the citric acid cycle [
59].
Ether phospholipids are particular classes of phospholipids containing a glycerol backbone with an ether or vinyl-ether bond at the
sn-1 position [
60]. Plasmalogen is one type of ether phospholipid containing a vinyl-ether bond that is widely found in the brain, heart, and white blood cells [
17]. The biosynthesis of plasmalogen begins in peroxisomes [
17,
60,
61]. Dihydroxyacetone (DHAP), a glycolysis intermediate, is used as a building block of the glycerol backbone of plasmalogen. The peroxisomal matrix enzyme glyceronephosphate
O-acyltransferase (GNPAT) transfers the acyl chain of acyl-CoA to DHAP to generate acyl-DHAP. Then, alkyl dihydroxyacetone phosphate synthase (AGPS) exchanges the acyl chain for an alkyl group to form alkyl-DHAP. Recently, Honsho et al. [
62] showed that acyl/alkyl-DHAP reductase (ADHAPR/DHRS7B) localizes in peroxisomes and the ER as a type I integral membrane protein in HeLa cells. The authors demonstrated that ER-localized ADHAPR reduces alkyl-DHAP, while peroxisomal ADHAPR preferentially reduces acyl-DHAP before the subsequent synthesis of alkyl-DHAP catalyzed by alkyl-DHAP synthase (ADAPS) [
62]. The remaining steps of plasmalogen synthesis take place in the ER [
63,
64].
Peroxisomes are also important for ROS metabolism. The name “peroxisome” was first introduced by De Duve in 1965 to define an organelle containing several enzymes that produce or break down hydrogen peroxide (H
2O
2) [
4,
65]. Peroxisomes are responsible for up to 20% of the cell’s oxygen consumption and 35% of the H
2O
2 production in mammalian tissues [
66]. Peroxisomes produce different ROS species such as superoxide, hydroxyl radicals, and hydrogen peroxide as byproducts of metabolic processes by multiple oxidases present in the organelle. Acyl-CoA oxidases (ACOX) are the most abundant ROS-generating enzymes in the peroxisome [
59]. D-amino acid oxidase (DAO), D-aspartate oxidase (DDO), L-pipecolic acid oxidase (PIPOX), L-α-hydroxy acid oxidases (HAO), polyamine oxidase (PAOX), and xanthine oxidase (XDH) are also important H
2O
2 producers in human peroxisomes. To protect the organelle from oxidative damage, human peroxisomes have several antioxidant enzymes including catalase (CAT), copper/zinc-containing superoxide dismutase (SOD1), peroxiredoxin 5 (PRDX5), glutathione S-transferase kappa 1 (GSTK1), microsomal GST1 (MGST1), and epoxide hydrolase 2 (EPHX2) [
67].
4. Cellular Responses to Peroxisomal Dysfunction
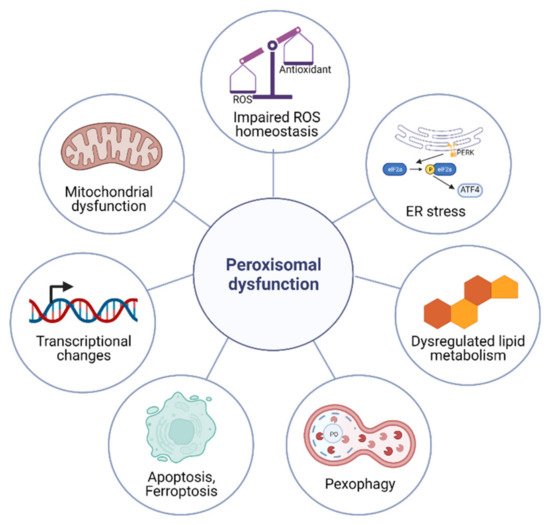
The metabolic functions of peroxisomes are critical for cellular homeostasis, and the impairment of proper peroxisomal function elicits a variety of cellular responses (Figure 1).
Figure 1. Cellular responses to peroxisomal dysfunction. Impaired peroxisomal function elicits cellular responses such as the impaired ROS homeostasis, dysregulated lipid metabolism, mitochondrial dysfunction, altered gene expression, pexophagy, elevated ER stress, and apoptosis.
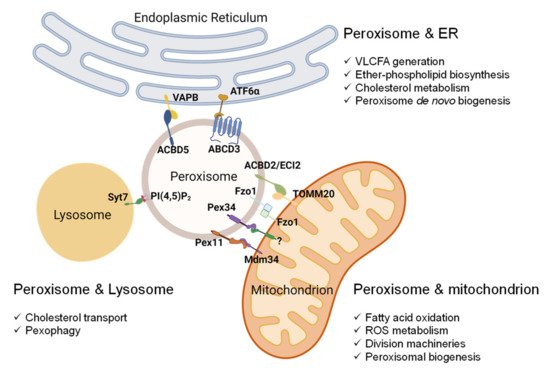
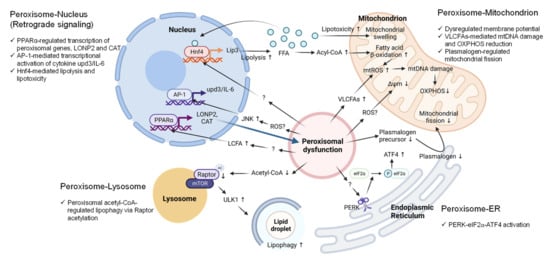
5. Peroxisome-Organelle Communication
Peroxisomes are multifunctional and dynamic organelles that actively contribute to cellular signaling, cell fate, immunity and inflammation, and aging [
6]. To accomplish these cellular activities, peroxisomes interact and coordinate with other subcellular organelles, such as mitochondria, ER, lysosomes, and lipid droplets [
6,
59,
104]. The cellular changes that accompany peroxisomal dysfunction are strongly associated with peroxisome-organelle crosstalk. Here, the current knowledge on peroxisome-organelle communication involving membrane contact sites, peroxisome-derived signaling metabolites, and retrograde signaling is summarized (
Figure 2 and
Figure 3).
Figure 2. Peroxisome-organelle membrane contact sites and their roles in organelle functions. Peroxisomes’ contact with mitochondria involves (1) peroxisomal Pex11 and mitochondrial Mdm34, (2) peroxisomal Pex34 and an unknown mitochondrial protein, (3) mitofusin Fzo1 on both peroxisome and mitochondria, and (4) peroxisomal ACBD2/ECI2 and mitochondrial TOMM20. The interaction between peroxisomes and the ER involves (1) peroxisomal ACBD5 and resident ER protein VAPB and (2) peroxisomal ABCD3 and ER protein ATF6α upon Ceapin treatment. In mammalian cells, the contact between peroxisomes and lysosomes involves phospholipid PI(4,5)P2 on the peroxisomal membrane and lysosomal Syt7. Abbreviations: Pex11, peroxin 11; Mdm34, mitochondrial distribution and morphology protein 34; Pex34, peroxin 34; Fzo1, mitofusin; ACBD2/ECI2, acyl-coenzyme A-binding domain or enoyl-CoA-δ isomerase 2; TOMM20, translocase of outer mitochondrial membrane 20; ACBD5, acyl-CoA binding domain-containing protein 5; VAPB, vesicle-associated membrane protein-associated protein B/C; ABCD3, ATP binding cassette subfamily D member 3; ATF6, activating transcription factor 6; PI(4,5)P2, phosphatidylinositol 4,5-bisphosphate; Syt7, synaptotagmin VII.
Figure 3. Metabolic and molecular signaling pathways mediate peroxisome-organelle crosstalk in response to peroxisomal dysfunction. (1) Peroxisome-nucleus: Peroxisomal dysfunction activates nuclear gene transcription through the transcription factors PPARα, AP-1, or Hnf4. The accumulation of long-chain fatty acids (LCFAs) activates PPARα to upregulate LONP2 and CAT expression to restore peroxisomal function (Blue arrow). Elevated ROS activates JNK signaling and AP-1 to induce the transcription of the pro-inflammatory cytokine IL-6. Peroxisomal dysfunction activates Hnf4 through unknown mechanisms to induce the expression of lipase 3 (Lip3), which leads to excessive free fatty acids (FFA) in the cytoplasm. FFAs hyper-activate Hnf4 via a positive feedback loop. (2) Peroxisome-mitochondrion: Peroxisomal dysfunction causes mitochondrial swelling, mitochondrial ROS generation, mitochondrial DNA (mtDNA) damage, disruption of oxidative phosphorylation (OXPHOS), and decreased inner membrane potential. Excessive ROS and elevated VLCFA are the potential signaling molecules that mediate the peroxisome-mitochondrion crosstalk. Peroxisomal dysfunction decreases plasmalogen, resulting in the inhibition of mitochondrial fission. (3) Peroxisome-ER: Peroxisomal dysfunction activates PERK signaling through unknown mechanisms to increase the phosphorylation of eIF2α and ATF4 induction. (4) Peroxisome-lysosome: Peroxisomal dysfunction due to the loss of Acox1 decreases peroxisome-derived acetyl-CoA, resulting in Raptor acetylation and lysosome localization of mTOR, leading to ULK1 activation and elevated lipophagy. Abbreviations: PPARα, peroxisome proliferator activated receptor alpha; AP-1, activator protein-1; Hnf4, hepatocyte nuclear factor 4; LONP2, lon peptidase 2; CAT, catalase; upd3, unpaired 3; IL-6, interleukin 6; Lip3, lipase 3; ROS, reactive oxygen species; JNK, Jun N-terminal kinase; VLCFAs, very-long-chain fatty acids; PERK, protein kinase RNA-like endoplasmic reticulum kinase; eIF2α, eukaryotic translation initiation factor 2a, ATF4, activating transcription factor 4; mTOR, mechanistic target of rapamycin kinase; ULK1, Unc-51-like autophagy-activating kinase 1.
6. Peroxisomal Dysfunction in Aging and Aging-Related Diseases
Several lines of evidence suggest that peroxisomal dysfunction is an underappreciated cause of aging [
11,
12,
13,
14]. Peroxisomal protein import is known to be compromised in aged tissues [
11,
12,
13,
14], and almost all peroxisomal proteins are downregulated during aging [
8,
13]. In senescent human fibroblasts, catalase import was significantly reduced, leading to the accumulation of H
2O
2 and the further disruption of peroxisome import [
11]. Conversely, the overexpression of
Pex5 has been shown to restore peroxisomal import function and preserve cardiac health in aged oenocytes [
14]. Importantly, Sebastiani et al. [
169] studied the proteomic signatures of centenarians and identified that proteins encoding components of peroxisomes were significantly downregulated in centenarians’ serum compared with younger individuals. In addition, the bile acid metabolism pathway and cholesterol homeostasis pathway were also downregulated in centenarians. These studies provide strong evidence for a role of impaired peroxisomal homeostasis in aging.
As peroxisomal activities decline with age [
170], it has been suggested that peroxisomal dysfunctions might be associated with the pathogenesis of age-related neurodegenerative disease, including Alzheimer’s disease [
171], Parkinson’s disease [
172], and amyotrophic lateral sclerosis [
173]), as well as age-related metabolic disease, such as cardiovascular disease [
174], obesity [
76], diabetes [
175], and nonalcoholic liver disease [
176]. These age-related pathologies are likely due to increased oxidative stress [
177,
178] and decreased plasmalogen synthesis [
179]. In this section, we summarize the current knowledge about the role of peroxisomes in several age-related diseases.
Alzheimer’s disease (AD) is the most common neurodegenerative disease in the elderly. It is characterized by the progressive loss of synapses and neurons especially in the frontal cortex and hippocampus accompanied by significant memory loss and cognitive impairment [
180,
181]. The pathological hallmarks of AD are the accumulation of the extracellular beta-amyloid (Aβ) plaques and intracellular fibrillary deposits of hyperphosphorylated tau protein (neurofibrillary tangles). Oxidative stress is thought to be a primary culprit in AD pathogenesis [
182,
183]. Accordingly, the hippocampus of AD mice was found to exhibit high levels of markers of lipid peroxidation and DNA/RNA oxidation [
182]. Several lines of evidence suggest a link between peroxisomal dysfunction and AD pathogenesis. The accumulation of VLCFAs and decreased plasmalogen levels were observed in the cortical regions of AD patients [
184]. Also, the levels of DHA in the brain and liver tissue of AD patients were reduced compared to those in control individuals [
185]. Impaired peroxisomal function might also contribute to alterations of peroxisomal metabolites in AD pathology. Shi et al. [
186] showed that the inhibition of peroxisomal β-oxidation increased the level of VLCFAs and Aβ contents in rat cerebral cortex. On the other hand, treatment with peroxisome proliferators attenuated AD-related pathology and improved spatial memory in transgenic mice models of AD [
187]. Although peroxisomal dysfunction is associated with AD pathogenesis, whether decreased peroxisomal activity is the primary cause, bystander, or consequence in AD pathology remains to be determined [
188].
Cardiovascular diseases (CVDs) are the major cause of death in elderly populations [
189], and the prevalence of CVDs has been shown to increase with age [
190]. Aging plays a critical role in the impairment of the cardiovascular system and increases the risk of CVDs [
191]. Peroxisomal dysfunction has been implicated in CVDs. For instance, cardiac-specific overexpression of catalase, predominantly located in peroxisomes [
192], was found to prevent the progression to overt heart failure [
174]. In addition, catalase activity is significantly decreased in failing myocardium [
193]. Refsum’s disease is a peroxisomal disorder caused by the impaired α-oxidation of branched-chain fatty acids, and patients with Refsum’s disease develop cardiac arrhythmia and heart failure later in life [
194,
195,
196]. We recently demonstrated that peroxisomal dysfunction in
Drosophila liver induced cardiac arrhythmia through the production of inflammatory cytokine
upd3/IL-6 [
14]. We showed that the production of
upd3/IL-6 activated the cardiac JAK-STAT signaling pathway and induced cardiac arrhythmia.
Aging is accompanied by changes in the distribution and composition of adipose tissue, particularly an increase in fat deposition within the white adipose tissue (abdominal obesity), which is a major contributor to insulin resistance and metabolic syndrome [
197,
198,
199]. As life expectancy increases, the prevalence of obesity has also risen steadily among the elderly [
199]. A recent study demonstrated that peroxisomes play an important role in adipose dysfunction associated with obesity [
76]. The authors found that obese mice exhibited downregulation of peroxisomal genes in white adipose tissue, and the knockdown of
Pex5 increased ROS levels and inflammation in adipocytes. Accordingly, catalase knockout mice also showed accelerated obesity compared to control mice. These phenotypes were attenuated by improving peroxisomal biogenesis through treatment with the PPARα agonist fenofibrate [
76]. Additionally, catalase-deficient patients frequently show diabetic pathology, presumably due to the accumulation of oxidative damage in pancreatic β-cells [
200].
This entry is adapted from the peer-reviewed paper 10.3390/antiox11020192