Protein tyrosine phosphatase gamma (PTPRG) is an enzyme that remove phosphate groups from phosphotyrosine residues of specific intracellular targets. It belongs to the tyrosine phosphatase (PTP) family of proteins and it is widely expressed in human tissues.
- Tyrosine phosphatase
- cell differentiation
- diabetes
- CNS disease
- liver disease
- kidney disease
- cell adhesion
- plasma
- endothelium
- leukemia
Introduction
Phosphorylation and dephosphorylation are important post-translational modifications that occur in the different aminoacid residues. These biological processes are essential mechanisms for intracellular signal transduction and for switching enzyme activity. The balance between the activities of protein tyrosine kinase (PTK) and protein tyrosine phosphatase (PTP) allows them to maintain cellular homeostasis [1]. PTPs are generally divided in proteins tyrosine phosphatase (PTPs) and proteins serine-threonine phosphatase (PSTPs) based on their substrate specificity for tyrosine, serine, and threonine [2][3]. Members of the PTP family are primarily involved in signaling pathways such as cell cycle regulation, proliferation, invasion and angiogenesis, acting as a natural counterpart of tyrosine kinases (TKs). Protein tyrosine phosphatase gamma (PTPRG) belongs to R5 group of the receptor type phosphatase subfamily (RPTP), together with its homologous PTPRZ, and is widely expressed in various isoforms in different tissues [4][5]. PTPRG gene encodes for a transmembrane protein composed in the extracellular region (ECD) by the homologous α-carbonic anhydrase like domain (CAH) and the Fibronectin type III domain and in the intracellular side by two phosphatase domains. The domain D1 is the catalytically active phosphatase portion while the domain D2 is a pseudophosphatase domain lacking of enzymatic activity and probably involved in the stability, substrate specificity and binding of docking proteins [6][7][8]. Several PTPRG isoforms have been described: PTPRG-A is a full-length isoform, PTPRG-B lacks 29 amino acids in the juxtamembrane position, PTPRG-C contains only one phosphatase domain and PTPRG-D (sPTPRG) is a soluble isoform wich has lost all phosphatase activity [9]. PTPRG isoforms have important roles in various human tissues and diseases, including different types of cancer and inflammatory disorders.
Physiological roles of PTPRG
PTPRG Affects Cell Differentiation
PTPRG was originally identified in the central nervous system (CNS) [6], together with PTPRZ [10][11], where it plays a role in differentiation and neurogenesis. PTPRG mRNA and the different protein isoforms are expressed in neuronal and accessory cells, including sensory pyramidal cells, microglia and Purkinje cells [12][13][14]. It was reported, in a study conducted in chicken spinal cord, that PTPRG perturbation significantly reduce progenitor rates and the number of neuronal precursors, resulting in neuroepithelial hypoplasia. This reveals its potential role in the survival and localization/movement of motor neuron precursor (MNs) [12]. In this contest, PTPRG was suggested to interact with the WNT/β-catenin signaling and the modulation of β-catenin phosphorylation levels with the consequent influence of the TCF/LEF complex. Loss of function of PTPRG was associated to increase apoptosis and defective cell–cell adhesion in progenitors and precursors of MNs while gain-of-function causes suppression of Wnt/β-catenin-driven TCF signalling [15]. In murine embryonic stem cells, PTPRG was expressed in a limited timeframe, suggesting an accurate regulation of PTPs during cell differentiation. Of note, its downregulation in embryonic stem (ES) cells is associated to complete lack of hematopoietic colonies [8]. Altogether, these data suggest an important regulatory role in key processes affecting embryonal development in different cellular models and species.
Cell Adhesion
Regarding the potential role of PTPRG in cell adhesion, it is known that the growth factor pleiotropin (PTN) and contactin (CNTN1) are able to bind the extracellular domain (ECD) of the homolog PTPRZ [16] and these interactions can regulate various signals during neuronal development [17][18]. Additionally, some members of this family (CNTN3-6) may also be potential ligands for the CAH domain of PTPRG [19]. Indeed, structural and biochemical investigations have shown an interaction with CNTN3-6, in particular CNTN3, which forms complexes with PTPRG on the membranes of different rod photoreceptors of the murine retina [20]. Furthermore, the silencing of CNTN6 or PTPRG deficiency, both expressed in layer V of the cerebral cortex, alters motor coordination in mouse models during specific tests [12][21] suggesting the importance of the interaction among these partners and the role of PTPRG in the acquisition of correct motor function. A link with adhesion processes was also found in leukocytes. Integrins are a large family of heterodimeric cell surface receptors and drive cell-to-cell and cell-to-matrix adhesion and migration mechanisms [22]. In human primary monocytes, the PTPRG phosphatase activity, prevent integrin activation triggered by chemoattractants mediating rapid arrest in underflow adhesion assays. Importantly, PTPRG seems to inhibit the transition to high-affinity state of LFA-1, through JAK2 dephosphorylation [23]. The same approach was utilized in healthy B-lymphocytes compared to chronic lymphocytic leukemia cells (CLL). Activated PTPRG decreases the activity of JAK2 and BTK kinases involved in chemokine signaling to LFA-1 and VLA-4 Integrins activation [24][25].
PTPRG as HCO3- Sensor
PTPRG and PTPRZ have extracellular domains (ECDs) that are 35%–40% identical to the catalytic domains of human carbonic anhydrases (CAs), family of enzymes involved in the interconversion of HCO3- and CO2 [26]. The CAH-like domain of PTPRG-ECD lacks key histidine residues essential for catalytic activity [6], suggesting a non-catalytic role of this domain. A possible role in the regulation of acid–base balance was described in renal proximal tubules (PTs) in PTPRG –/– mice, emphasizing the loss of ability to counteract the basolateral [HCO3-] changes, compared to WT. In addition, in a simulated metabolic acidosis PTPRG interferes with the restoration of the arteriolar pH compared to WT, showing a substantial acidosis in PTPRG –/– mice [27]. In supporting the involvement of PTPRG as CO2/ HCO3- sensor, the same role was also confirmed at the arteriolar level in PTPRG –/– mice. Lastly, Ptprg was suggested to exert HCO3- sensor function and to regulate the endothelium-dependent vasorelaxant effects. In particular, it has demonstrated that endothelial Ptprg affects the vasomotor effects induced by CO2/HCO3- by regulating the Ca2+ sensitivity in arteriolar VSMC (vascular smooth muscle cells) that might enhances intracellular [Ca2+ ] in endothelial cells by implementing NO synthesis and endothelium-dependent hyperpolarization (EDH)-type responses [28][29].
PTPRG in disease
Cancers
PTPRG is considered a tumor-suppressor gene (TSG) mapped on chromosome 3p14-21, a region subjected to genetic and epigenetic alterations, including non-random deletions, loss of heterozygosity, and alteration of the CpG islands methylation profile in several types of cancer [30][31]. In particular, PTPRG gene shows promoter hypermethylation in cutaneous T-cell lymphoma and melanoma and it harbors point mutations and deletions region in colon, renal and lung carcinomas. Moreover, lower expression levels of PTPRG have been reported in various cancers [32][33][34]. However, a tumor suppressor role in tumors might not be ruled out. In particular, in Central Nervous System (CNS) PTPRG appears to be crucial in maintaining glioblastoma cell-related neuronal stemness, carving out a pathological functional role in this tissue [35].
Neuropsychiatric and behavioral disorders
In addition to the role of PTPs in the differentiation and synaptic organization of the CNS, they can take part during the modulation of the processes related to learning and memory [36][37]. Harroch’s group, using PTPRG–/– and PTPRZ–/– mice identified specific behavioral changes in the animals. The loss of combined or individual phosphatase was associated with a specific neurotransmitter modulation in distinct areas of the brain, indicating these as appealing therapeutic targets [38]. In the present case, PTPRG has a precise distribution in the brain and is characteristic of a group of neurons [12][13]. Its high affinity for contactin members, which are involved in autism spectrum disorder (ASD), indicate a potential role in this disease [39][40]. Several evidence suggesting PTPRG as a susceptible gene involved also in neuropsychiatric disorders. Whole-genome sequencing analysis in several cohorts of schizophrenic patients revealed mutations involving five genes, including PTPRG. Likewise, PTPRG has been associated with Alzheimer’s disease (AD), one of the most frequent forms of dementia in the world [41]. The predominant form of Alzheimer’s diagnosed is late onset (LOAD), in which the strongest risk factor considered remains APOE-"4. A single-nucleotide polymorphism (SNP) was identified on PTPRG; the rs7609954 variant achieves high genome-wide significance pointing to PTPRG and its phosphatase regulatory functions as a possible risk gene for late-onset forms of Alzheimer's disease [42].
Inflammation
The study of PTPRG in the CNS of rat brain unearthed different PTPRG isoforms derived from alternative splicing. Higher expression of the soluble protein form (sPTPRG) was found during the embryonic stages [43]. In mice treated with LPS for 24 h, a general inflammatory state was induced and was associated with increased expression of all PTPRG isoforms in cortical astrocytes, particularly the 120 KDa soluble isoform. In 5XFAD Alzheimer’s disease mouse model, astrocytes surrounding the -amyloid plaques are highly positive for PTPRG expression, suggesting as a possible factor for astrocytic activation during neuroinflammation [13]. Furthermore, this processed isoform (sPTPRG) has been shown to be present in both human and murine liver and serum. These data led to the analysis of primary serum samples from healthy donors and patients with elevated alanine aminotransferase (ALT), an established biomarker of liver injury [44]. High serum ALT levels correlate with increased levels of soluble PTPRG compared to healthy donor samples, suggesting PTPRG as a putative biomarker of liver injury [45]. Potential role as disease biomarker is suggested by the analysis of cerebrospinal fluid (CSF) that identified the closely related sPTPRZ as a biomarker of brain tumors [46]. PTPRG involvement in metabolic disorders was also supported by several published data. Chronic inflammatory processes are often found in overweight or obese individuals, where complications such as insulin resistance, type 2 diabetes (T2DM), liver and cardiovascular disease and cancer may occur [47][48]. In this context a role of phosphatases as insulin receptor (IR) inactivators was proposed [49]. In particular Bottini’s group identified small-molecule inhibitor of LMWPTP (low-molecular-weight tyrosine phosphatase) capable of blocking insulin resistance and T2DM occurrence [50]. In humans liver PTPRG overexpression appears to correlate with inflammatory mechanisms and obesity [51]. Molecular mechanisms analysis performed on PTPRG–/– mice showed a robust improvement in glucose metabolism induced by the increased insulin sensitivity in hepatocytes, thus remaining protected from the development of hyperglycemia when compared to PTPRG+/+. Hepatic insulin resistance was found in Ptprg-overexpressing mice at values comparable to obese subjects. Interestingly inflammatory states such as obesity are able to generate an NF-kB-induced PTPRG expression mechanism due to a kB-binding site identified on the PTPRG promoter [51].
Other Diseases
Several PTPRG roles are yet to be confirmed, but some recent evidence has linked this phosphatase with renal and corneal dysfunctions. Acute kidney injury (AKI) is a kidney disorder characterized by a heterogeneous group of dysfunctions such as decreased glomerular filtration rate with and oliguria with a variable etiology [52]. The study of biomarkers such as miRNAs in the various stages of AKI suggested a possible prediction profile of recovering/non-recovering patients after 90 days. A screening on different miRNAs in AKI patients has reported the upregulation of miR-141, which both in a model of H2O2-induced tissue damage or in samples disease is able to negatively modulate PTPRG mRNA expression. The downregulation of PTPRG could therefore be fundamental for the acceleration of renal fibrotic processes through EGFR hyper-stimulation, formerly known as PTPRG substrate [53]. Degenerative disease named Fuchs’ endothelial dystrophy (FED) can affect the corneal endothelium, generating microscopic outgrowths (guttae) in the corresponding basal lamina. Although a strong correlation with the disease was found for some genes (TCF4, TCF8, AGBL1, LOXHD1, SLC4A11, and others), the study of PTPRG on several SNPs (rs7640737 and rs10490775) appear conflicting, showing an association without reaching a genome-wide significance in FED [54][55][56][57].
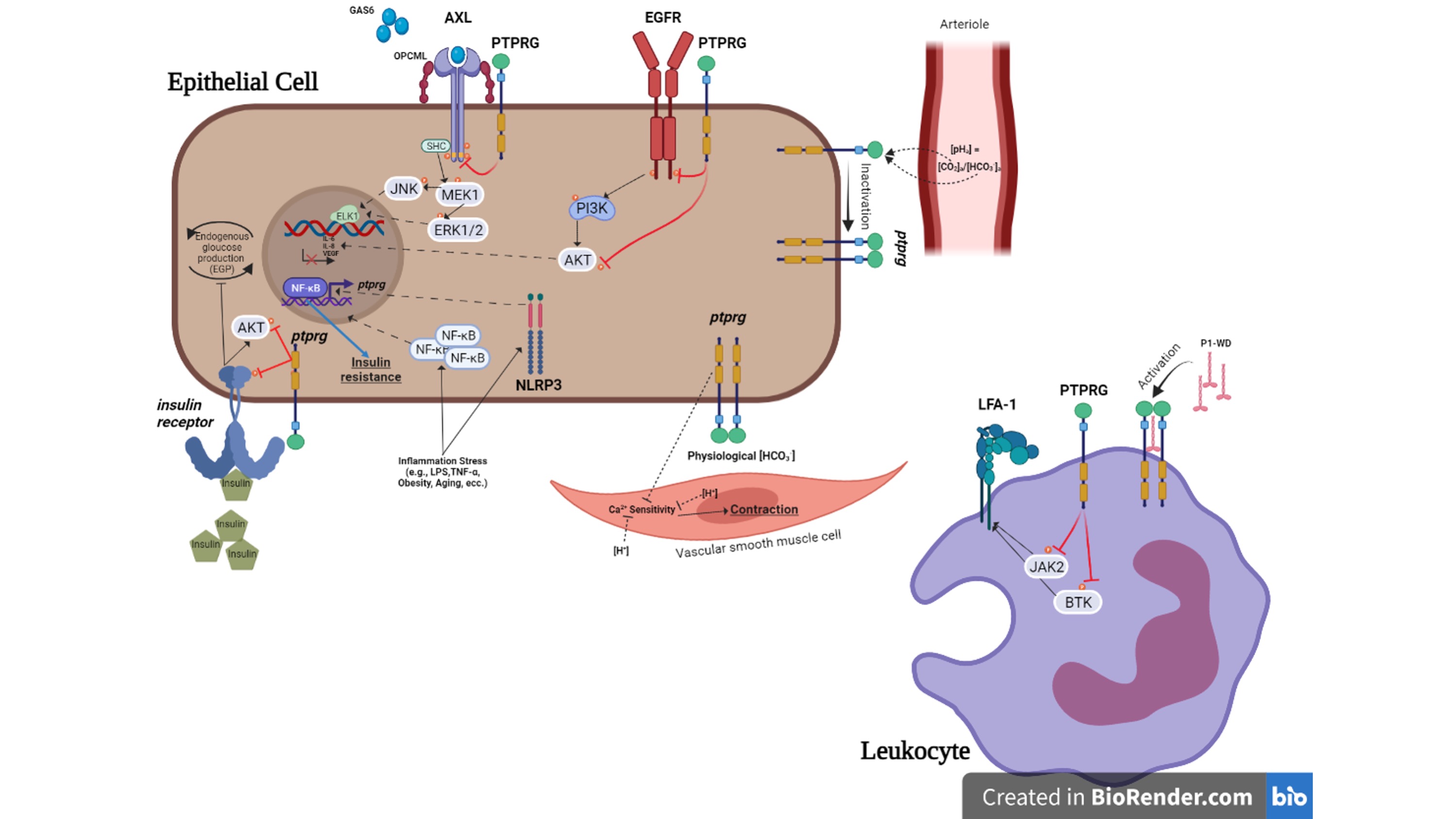
Figure 1. Summary of the role of PTPRG in physiopathology of non-neoplastic cell
References
- Liberti, S.; Sacco, F.; Calderone, A.; Perfetto, L.; Iannuccelli, M.; Panni, S.; Santonico, E.; Palma, A.; Nardozza, A.P.; Castagnoli, L.; et al. HuPho: The human phosphatase portal. FEBS J. 2013, 280, 379–387.
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846.
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484.
- Krueger, N.X.; Saito, H. A human transmembrane protein-tyrosine-phosphatase, PTP zeta, is expressed in brain and has an N-terminal receptor domain homologous to carbonic anhydrases. Proc. Natl. Acad. Sci. USA 1992, 89, 7417–7421.
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711.
- Barnea, G.; Silvennoinen, O.; Shaanan, B.; Honegger, A.M.; Canoll, P.D.; D’Eustachio, P.; Morse, B.; Levy, J.B.; Laforgia, S.; Huebner, K.; et al. Identification of a carbonic anhydrase-like domain in the extracellular region of RPTP gamma defines a new subfamily of receptor tyrosine phosphatases. Mol. Cell Biol. 1993, 13, 1497–1506.
- Tonks, N.K. Protein tyrosine phosphatases–from housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280, 346–378.
- Sorio, C.; Melotti, P.; D’Arcangelo, D.; Mendrola, J.; Calabretta, B.; Croce, C.M.; Huebner, K. Receptor protein tyrosine phosphatise gamma, Ptp gamma, regulates hematopoietic differentiation. Blood 1997, 90, 49–57.
- Jiang, G.; den Hertog, J.; Su, J.; Noel, J.; Sap, J.; Hunter, T. Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-alpha. Nature 1999, 401, 606–610.
- Levy, J.B.; Canoll, P.D.; Silvennoinen, O.; Barnea, G.; Morse, B.; Honegger, A.M.; Huang, J.T.; Cannizzaro, L.A.; Park, S.H.; Druck, T.; et al. The cloning of a receptor-type protein tyrosine phosphatase expressed in the central nervous system. J. Biol. Chem. 1993, 268, 10573–10581.
- Tamura, H.; Fukada, M.; Fujikawa, A.; Noda, M. Protein tyrosine phosphatase receptor type Z is involved in hippocampusdependent memory formation through dephosphorylation at Y1105 on p190 RhoGAP. Neurosci. Lett. 2006, 399, 33–38.
- Lamprianou, S.; Vacaresse, N.; Suzuki, Y.; Meziane, H.; Buxbaum, J.D.; Schlessinger, J.; Harroch, S. Receptor protein tyrosine phosphatase gamma is a marker for pyramidal cells and sensory neurons in the nervous system and is not necessary for normal development. Mol. Cell Biol. 2006, 26, 5106–5119.
- Lorenzetto, E.; Moratti, E.; Vezzalini, M.; Harroch, S.; Sorio, C.; Buffelli, M. Distribution of different isoforms of receptor protein tyrosine phosphatase gamma (Ptprg-RPTP gamma) in adult mouse brain: Upregulation during neuroinflammation. Brain Struct. Funct. 2014, 219, 875–890.
- Vezzalini, M.; Mombello, A.; Menestrina, F.; Mafficini, A.; Della Peruta, M.; van Niekerk, C.; Barbareschi, M.; Scarpa, A.; Sorio, C. Expression of transmembrane protein tyrosine phosphatase gamma (PTPgamma) in normal and neoplastic human tissues. Histopathology 2007, 50, 615–628.
- Hashemi, H.; Hurley, M.; Gibson, A.; Panova, V.; Tchetchelnitski, V.; Barr, A.; Stoker, A.W. Receptor tyrosine phosphatase PTPgamma is a regulator of spinal cord neurogenesis. Mol. Cell Neurosci. 2011, 46, 469–482.
- Fukada, M.; Fujikawa, A.; Chow, J.P.; Ikematsu, S.; Sakuma, S.; Noda, M. Protein tyrosine phosphatase receptor type Z is inactivated by ligand-induced oligomerization. FEBS Lett. 2006, 580, 4051–4056.
- Peles, E.; Nativ, M.; Campbell, P.L.; Sakurai, T.; Martinez, R.; Lev, S.; Clary, D.O.; Schilling, J.; Barnea, G.; Plowman, G.D.; et al. The carbonic anhydrase domain of receptor tyrosine phosphatase beta is a functional ligand for the axonal cell recognition molecule contactin. Cell 1995, 82, 251–260.
- Chatterjee, M.; Schild, D.; Teunissen, C.E. Contactins in the central nervous system: Role in health and disease. Neural Regen. Res.2019, 14, 206–216.
- Bouyain, S.; Watkins, D.J. The protein tyrosine phosphatases PTPRZ and PTPRG bind to distinct members of the contactin family of neural recognition molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 2443–2448.
- Nikolaienko, R.M.; Hammel, M.; Dubreuil, V.; Zalmai, R.; Hall, D.R.; Mehzabeen, N.; Karuppan, S.J.; Harroch, S.; Stella, S.L.; Bouyain, S. Structural Basis for Interactions Between Contactin Family Members and Protein-tyrosine Phosphatase Receptor Type G in Neural Tissues. J. Biol. Chem. 2016, 291, 21335–21349.
- Takeda, Y.; Akasaka, K.; Lee, S.; Kobayashi, S.; Kawano, H.; Murayama, S.; Takahashi, N.; Hashimoto, K.; Kano, M.; Asano, M.; et al. Impaired motor coordination in mice lacking neural recognition molecule NB-3 of the contactin/F3 subgroup. J. Neurobiol.2003, 56, 252–265.
- Nolte, M.A.; Margadant, C. Activation and suppression of hematopoietic integrins in hemostasis and immunity. Blood 2020, 135,7–16.
- Mirenda, M.; Toffali, L.; Montresor, A.; Scardoni, G.; Sorio, C.; Laudanna, C. Protein tyrosine phosphatase receptor type gamma is a JAK phosphatase and negatively regulates leukocyte integrin activation. J. Immunol. 2015, 194, 2168–2179.
- Montresor, A.; Toffali, L.; Fumagalli, L.; Constantin, G.; Rigo, A.; Ferrarini, I.; Vinante, F.; Laudanna, C. Activation of Protein Tyrosine Phosphatase Receptor Type gamma Suppresses Mechanisms of Adhesion and Survival in Chronic Lymphocytic Leukemia Cells. J. Immunol. 2021, 207, 671–684.
- Montresor, A.; Toffali, L.; Rigo, A.; Ferrarini, I.; Vinante, F.; Laudanna, C. CXCR4- and BCR-triggered integrin activation in B-cell chronic lymphocytic leukemia cells depends on JAK2-activated Bruton’s tyrosine kinase. Oncotarget 2018, 9, 35123–35140.
- Aspatwar, A.; Tolvanen, M.E.; Ortutay, C.; Parkkila, S. Carbonic anhydrase related proteins: Molecular biology and evolution.Subcell. Biochem. 2014, 75, 135–156. [CrossRef] [PubMed]
- Zhou, Y.; Skelton, L.A.; Xu, L.; Chandler, M.P.; Berthiaume, J.M.; Boron, W.F. Role of Receptor Protein Tyrosine Phosphatase gamma in Sensing Extracellular CO2 and HCO3. J. Am. Soc. Nephrol. 2016, 27, 2616–2621.
- Boedtkjer, E.; Hansen, K.B.; Boedtkjer, D.M.B.; Aalkjaer, C.; Boron, W.F. Extracellular HCO3- is sensed by mouse cerebral arteries: Regulation of tone by receptor protein tyrosine phosphatase gamma. J. Cereb. Blood Flow Metab. 2016, 36, 965–980.
- Hansen, K.B.; Staehr, C.; Rohde, P.D.; Homilius, C.; Kim, S.; Nyegaard, M.; Matchkov, V.V.; Boedtkjer, E. PTPRG is an ischemia risk locus essential for HCO3- dependent regulation of endothelial function and tissue perfusion. Elife 2020, 9, e57553.
- Boni, C.; Sorio, C. The role of the tumor suppressor gene Protein Tyrosine Phosphatase Gamma (PTPRG) in cancer. Front. Cell Dev. Biol. 2021.
- Druck, T.; Kastury, K.; Hadaczek, P.; Podolski, J.; Toloczko, A.; Sikorski, A.; Ohta, M.; LaForgia, S.; Lasota, J.; McCue, P.; et al. Loss of heterozygosity at the familial RCC t(3;8) locus in most clear cell renal carcinomas. Cancer Res. 1995, 55, 5348–5353.
- LaForgia, S.; Morse, B.; Levy, J.; Barnea, G.; Cannizzaro, L.A.; Li, F.; Nowell, P.C.; Boghosian-Sell, L.; Glick, J.;Weston, A.; et al. Receptor protein-tyrosine phosphatase gamma is a candidate tumor suppressor gene at human chromosome region 3p21. Proc. Natl. Acad. Sci. USA 1991, 88, 5036–5040.
- Wang, Z.; Shen, D.; Parsons, D.W.; Bardelli, A.; Sager, J.; Szabo, S.; Ptak, J.; Silliman, N.; Peters, B.A.; van der Heijden, M.S.; et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 2004, 304, 1164–1166.
- Shu, S.T.; Sugimoto, Y.; Liu, S.; Chang, H.L.; Ye, W.; Wang, L.S.; Huang, Y.W.; Yan, P.; Lin, Y.C. Function and regulatory mechanisms of the candidate tumor suppressor receptor protein tyrosine phosphatase gamma (PTPRG) in breast cancer cells. Anticancer Res. 2010, 30, 1937–1946.
- Fujikawa, A.; Sugawara, H.; Tanaka ,T.; Matsumoto , M.; Kuboyama, K.; Suzuki, R.; Tanga ,N.; Ogata, N.; Masumura, M.; Noda, M. Targeting PTPRZ inhibits stem cell-like properties and tumorigenicity in glioblastoma cells. Sci Rep. 2017 Jul 17;7(1):5609.
- Le, H.T.; Maksumova, L.; Wang, J.; Pallen, C.J. Reduced NMDA receptor tyrosine phosphorylation in PTPalpha-deficient mouse synaptosomes is accompanied by inhibition of four src family kinases and Pyk2: An upstream role for PTPalpha in NMDA receptor regulation. J. Neurochem. 2006, 98, 1798–1809.
- Takahashi, H.; Craig, A.M. Protein tyrosine phosphatases PTPdelta, PTPsigma, and LAR: Presynaptic hubs for synapse organization.Trends Neurosci. 2013, 36, 522–534.
- Cressant, A.; Dubreuil, V.; Kong, J.; Kranz, T.M.; Lazarini, F.; Launay, J.M.; Callebert, J.; Sap, J.; Malaspina, D.; Granon, S.; et al. Loss-of-function of PTPR gamma and zeta, observed in sporadic schizophrenia, causes brain region-specific deregulation of monoamine levels and altered behavior in mice. Psychopharmacology 2017, 234, 575–587.
- Gandawijaya, J.; Bamford, R.A.; Burbach, J.P.H.; Oguro-Ando, A. Cell Adhesion Molecules Involved in Neurodevelopmental Pathways Implicated in 3p-Deletion Syndrome and Autism Spectrum Disorder. Front. Cell Neurosci. 2020, 14, 611379.
- Zuko, A.; Kleijer, K.T.E.; Oguro-Ando, A.; Kas, M.J.H.; van Daalen, E.; van der Zwaag, B.; Burbach, J.P.H. Contactins in the neurobiology of autism. Eur. J. Pharmacol. 2013, 719, 63–74.
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031.
- Herold, C.; Hooli, B.V.; Mullin, K.; Liu, T.; Roehr, J.T.; Mattheisen, M.; Parrado, A.R.; Bertram, L.; Lange, C.; Tanzi, R.E. Familybased association analyses of imputed genotypes reveal genome-wide significant association of Alzheimer’s disease with OSBPL6, PTPRG, and PDCL3. Mol. Psychiatry 2016, 21, 1608–1612.
- Shintani, T.; Maeda, N.; Nishiwaki, T.; Noda, M. Characterization of rat receptor-like protein tyrosine phosphatase gamma isoforms. Biochem. Biophys. Res. Commun. 1997, 230, 419–425. [CrossRef] EMBO J. 1992, 11, 897–907.
- Eguchi, A.;Wree, A.; Feldstein, A.E. Biomarkers of liver cell death. J. Hepatol. 2014, 60, 1063–1074.
- Moratti, E.; Vezzalini, M.; Tomasello, L.; Giavarina, D.; Sorio, C. Identification of protein tyrosine phosphatase receptor gamma extracellular domain (sPTPRG) as a natural soluble protein in plasma. PLoS ONE 2015, 10, e0119110.
- Yamanoi, Y.; Fujii, M.; Murakami, Y.; Nagai, K.; Hoshi, K.; Hashimoto, Y.; Honda, T.; Saito, K.; Kitazume, S. Soluble protein tyrosine phosphatase receptor type Z (PTPRZ) in cerebrospinal fluid is a potential diagnostic marker for glioma. Neurooncol. Adv. 2020, 2, vdaa055.
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4.
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin. Investig. 2017, 127, 43–54.
- Crunkhorn, S. Metabolic Disease: Protein tyrosine phosphatase inhibitor reverses diabetes. Nat. Rev. Drug Discov. 2017, 16, 312–313.
- Stanford, S.M.; Aleshin, A.E.; Zhang, V.; Ardecky, R.J.; Hedrick, M.P.; Zou, J.; Ganji, S.R.; Bliss, M.R.; Yamamoto, F.; Bobkov, A.A.; et al. Diabetes reversal by inhibition of the low-molecular-weight tyrosine phosphatase. Nat. Chem. Biol. 2017, 13, 624–632.
- Brenachot, X.; Ramadori, G.; Ioris, R.M.; Veyrat-Durebex, C.; Altirriba, J.; Aras, E.; Ljubicic, S.; Kohno, D.; Fabbiano, S.; Clement, S.; et al. Hepatic protein tyrosine phosphatase receptor gamma links obesity-induced inflammation to insulin resistance. Nat. Commun. 2017, 8, 1820.
- Levey, A.S.; James, M.T. Acute Kidney Injury. Ann. Intern Med. 2018, 168, 837.
- Levey, A.S.; James, M.T. Acute Kidney Injury. Ann. Intern Med. 2018, 168, 837. Newbury, L.J.; Simpson, K.; Khalid, U.; John, I.; de Rivera, L.B.; Lu, Y.A.; Lopez-Anton, M.;Watkins,W.J.; Jenkins, R.H.; Fraser, D.J.; et al. miR-141 mediates recovery from acute kidney injury. Sci. Rep. 2021, 11, 16499.
- Baratz, K.H.; Tosakulwong, N.; Ryu, E.; Brown, W.L.; Branham, K.; Chen, W.; Tran, K.D.; Schmid-Kubista, K.E.; Heckenlively, J.R.; Swaroop, A.; et al. E2-2 protein and Fuchs’s corneal dystrophy. N. Engl. J. Med. 2010, 363, 1016–1024.
- Kuot, A.; Hewitt, A.W.; Griggs, K.; Klebe, S.; Mills, R.; Jhanji, V.; Craig, J.E.; Sharma, S.; Burdon, K.P. Association of TCF4 and CLU polymorphisms with Fuchs’ endothelial dystrophy and implication of CLU and TGFBI proteins in the disease process. Eur. J. Hum. Genet. 2012, 20, 632–638.
- Lau, L.C.; Ma, L.; Young, A.L.; Rong, S.S.; Jhanji, V.; Brelen, M.E.; Pang, C.P.; Chen, L.J. Association of common variants in TCF4 and PTPRG with Fuchs’ corneal dystrophy: A systematic review and meta-analysis. PLoS ONE 2014, 9, e109142.
- Wang, K.J.; Jhanji, V.; Chen, J.; Law, R.W.; Leung, A.T.; Zhang, M.;Wang, N.; Pang, C.P.; Yam, G.H. Association of transcription factor 4 (TCF4) and protein tyrosine phosphatase, receptor type G (PTPRG) with corneal dystrophies in southern Chinese. Ophthalmic Genet. 2014, 35, 138–141.