Calcium signaling plays important roles in physiological and pathological conditions, including cutaneous melanoma, the most lethal type of skin cancer. Intracellular calcium concentration ([Ca2+]i), cell membrane calcium channels, calcium related proteins (S100 family, E-cadherin, and calpain), and Wnt/Ca2+ pathways are related to melanogenesis and melanoma tumorigenesis and progression. Calcium signaling influences the melanoma microenvironment, including immune cells, extracellular matrix (ECM), the vascular network, and chemical and physical surroundings. Other ionic channels, such as sodium and potassium channels, are engaged in calcium-mediated pathways in melanoma. Calcium signaling serves as a promising pharmacological target in melanoma treatment, and its dysregulation might serve as a marker for melanoma prediction.
- calcium
- melanoma
- progression
- melanoma microenvironment
- mitochondria
1. Calcium Signaling in Melanoma Progression
1.1. [Ca2+]i Oscillation Influences Melanoma Progression
1.2. Calcium Channels Are Involved in Melanoma Progression
1.3. Ca2+ Signaling Influences Melanoma Progression through the Change of Morphological and Phenotypical Changes
1.4. Calcium-Related Pathways Participate in Melanoma Progression
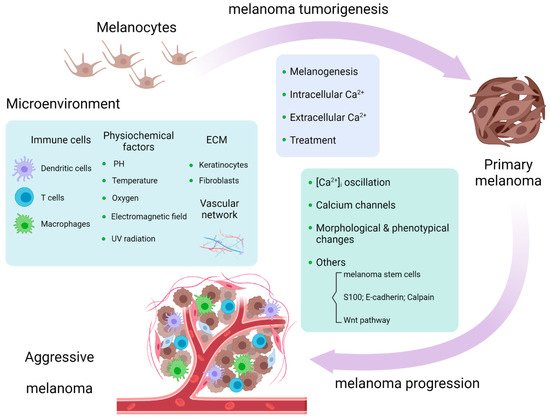
2. Calcium Signaling in Melanoma Microenvironment
2.1. Immune Cells
2.2. ECM and Vascular Network
2.3. Physical and Chemical Surroundings
This entry is adapted from the peer-reviewed paper 10.3390/ijms23031010