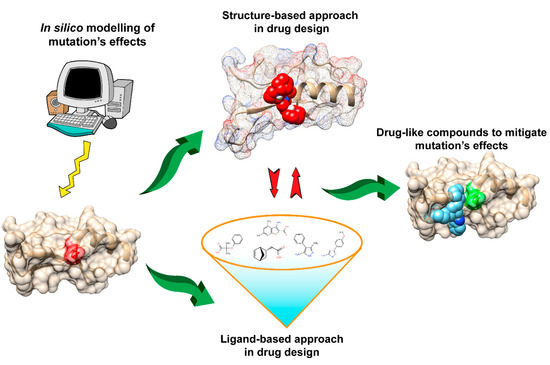
Structure-based drug design (SBDD) is the computational approach that relies on knowledge of 3D structures of the biological targets to identify or design the potential chemical structure suitable for clinical tests. With the explosion of genomic, functional, and structural information in recent decades, the majority of biological targets with 3D structure have been identified and stimulated the applications of structure-based approaches in the current design pipeline. SBDD is popular for virtual screening to filter the drug-like compounds from a large library of small molecules, including widely applied approaches, such as docking and structure-based pharmacophore design.
- mutations
- drug design
- macro-molecular docking
- pharmacophores
1. Docking
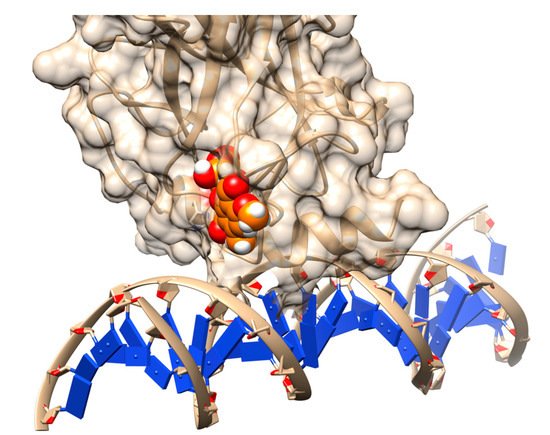
2. Structure-Based Pharmacophore Design
This entry is adapted from the peer-reviewed paper 10.3390/ijms20030548
References
- Shoichet, B.K.; Baase, W.A.; Kuroki, R.; Matthews, B.W. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. USA 1995, 92, 452–456.
- Pepys, M.B.; Hawkins, P.N.; Booth, D.R.; Vigushin, D.M.; Tennent, G.A.; Soutar, A.K.; Totty, N.; Nguyen, O.; Blake, C.C.F.; Terry, C.J.; et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature 1993, 362, 553–557.
- Hartley, R.W. Directed mutagenesis and barnase-barstar recognition. Biochemistry 1993, 32, 5978–5984.
- Buckle, A.M.; Schreiber, G.; Fersht, A.R. Protein-protein recognition: Crystal structural analysis of a barnase-barstar complex at 2.0-A resolution. Biochemistry 1994, 33, 8878–8889.
- Hung, C.-L.; Chen, C.-C. Computational approaches for drug discovery. Drug Dev. Res. 2014, 75, 412–418.
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749.
- Jain, A.N. Surflex: Fully Automatic Flexible Molecular Docking Using a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003, 46, 499–511.
- Basu, S.; Bhattacharyya, D.; Banerjee, R. Mapping the distribution of packing topologies within protein interiors shows predominant preference for specific packing motifs. BMC Bioinf. 2011, 12, 195.
- Michel, J. Current and emerging opportunities for molecular simulations in structure-based drug design. Phys. Chem. Chem. Phys. 2014, 16, 4465–4477.
- Nair, P.C.; Miners, J.O. Molecular dynamics simulations: From structure function relationships to drug discovery. In Silico Pharmacol 2014, 2, 4.
- Banerjee, R.; Sen, M.; Bhattacharya, D.; Saha, P. The jigsaw puzzle model: Search for conformational specificity in protein interiors. J. Mol. Biol. 2003, 333, 211–226.
- Charneski, C.A.; Hurst, L.D. Positive charge loading at protein termini is due to membrane protein topology, not a translational ramp. Mol. Biol. Evol. 2014, 31, 70–84.
- Harley, C.A.; Tipper, D.J. The Role of Charged Residues in Determining Transmembrane Protein Insertion Orientation in Yeast. J. Biol. Chem. 1996, 271, 24625–24633.
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310.
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53.
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317.
- Bullock, A.N.; Fersht, A.R. Rescuing the function of mutant p53. Nat. Rev. Cancer 2001, 1, 68–76.
- Coskuner-Weber, O.; Uversky, V.N. Insights into the Molecular Mechanisms of Alzheimer’s and Parkinson’s Diseases with Molecular Simulations: Understanding the Roles of Artificial and Pathological Missense Mutations in Intrinsically Disordered Proteins Related to Pathology. Int. J. Mol. Sci. 2018, 19.
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.-K.; Hall, L.V.; Salehi, F.; Lin, D.-W.; Chung, B.P.; Wesley Hatfield, G.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407.
- Kaar, J.L.; Basse, N.; Joerger, A.C.; Stephens, E.; Rutherford, T.J.; Fersht, A.R. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010, 19, 2267–2278.
- Pegg, A.E.; Michael, A.J. Spermine synthase. Cell. Mol. Life Sci. 2010, 67, 113–121.
- Peng, Y.; Norris, J.; Schwartz, C.; Alexov, E. Revealing the Effects of Missense Mutations Causing Snyder-Robinson Syndrome on the Stability and Dimerization of Spermine Synthase. Int. J. Mol. Sci. 2016, 17, 77.
- Zhang, Z.; Martiny, V.; Lagorce, D.; Ikeguchi, Y.; Alexov, E.; Miteva, M.A. Rational Design of Small-Molecule Stabilizers of Spermine Synthase Dimer by Virtual Screening and Free Energy-Based Approach. PLoS ONE 2014, 9, e110884.
- Zhang, Z.; Witham, S.; Petukh, M.; Moroy, G.; Miteva, M.; Ikeguchi, Y.; Alexov, E. A rational free energy-based approach to understanding and targeting disease-causing missense mutations. J. Am. Med. Inform. Assoc. 2013, 20, 643–651.
- Dror, O.; Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. Novel approach for efficient pharmacophore-based virtual screening: Method and applications. J. Chem. Inf. Model. 2009, 49, 2333–2343.
- Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules 2015, 20, 22799–22832.
- Lee, C.-H.; Huang, H.-C.; Juan, H.-F. Reviewing ligand-based rational drug design: The search for an ATP synthase inhibitor. Int. J. Mol. Sci. 2011, 12, 5304–5318.
- Chen, Z.; Li, H.; Zhang, Q.; Bao, X.; Yu, K.; Luo, X.; Zhu, W.; Jiang, H. Pharmacophore-based virtual screening versus docking-based virtual screening: A benchmark comparison against eight targets. Acta Pharmacol. Sin. 2009, 30, 1694–1708.
- Singh, P.K.; Silakari, O. Molecular dynamics guided development of indole based dual inhibitors of EGFR (T790M) and c-MET. Bioorg. Chem. 2018, 79, 163–170.
- Springsteel, M.F.; Galietta, L.J.V.; Ma, T.; By, K.; Berger, G.O.; Yang, H.; Dicus, C.W.; Choung, W.; Quan, C.; Shelat, A.A.; et al. Benzoflavone activators of the cystic fibrosis transmembrane conductance regulator: Towards a pharmacophore model for the nucleotide-binding domain. Bioorg. Med. Chem. 2003, 11, 4113–4120.
- Pathak, D.; Chadha, N.; Silakari, O. Identification of non-resistant ROS-1 inhibitors using structure based pharmacophore analysis. J. Mol. Graph. Model. 2016, 70, 85–93.
- Wang, A.; Li, X.; Wu, H.; Zou, F.; Yan, X.-E.; Chen, C.; Hu, C.; Yu, K.; Wang, W.; Zhao, P.; et al. Discovery of (R)-1-(3-(4-Amino-3-(3-chloro-4-(pyridin-2-ylmethoxy)phenyl)-1H-pyrazolo[3,4-d]pyrimidin-1-yl)piperidin-1-yl)prop-2-en-1-one (CHMFL-EGFR-202) as a Novel Irreversible EGFR Mutant Kinase Inhibitor with a Distinct Binding Mode. J. Med. Chem. 2017, 60, 2944–2962.
- Bleicher, K.H.; Böhm, H.-J.; Müller, K.; Alanine, A.I. Hit and lead generation: Beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378.
- Goldstraw, P.; Ball, D.; Jett, J.R.; Le Chevalier, T.; Lim, E.; Nicholson, A.G.; Shepherd, F.A. Non-small-cell lung cancer. Lancet 2011, 378, 1727–1740.
- Awad, M.M.; Katayama, R.; McTigue, M.; Liu, W.; Deng, Y.-L.; Brooun, A.; Friboulet, L.; Huang, D.; Falk, M.D.; Timofeevski, S.; et al. Acquired Resistance to Crizotinib from a Mutation in CD74–ROS1. N. Engl. J. Med. 2013, 368, 2395–2401.