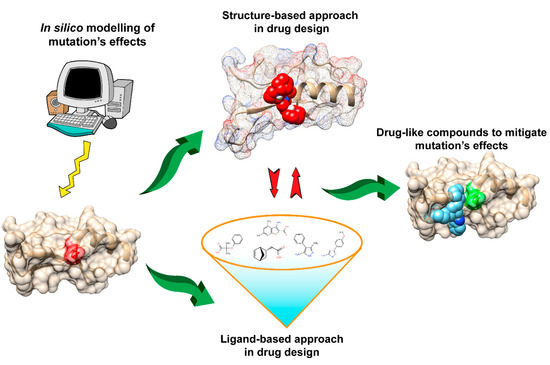
Docking is one common approach for compound screening. The basic idea is to use scoring functions [
1,
2,
3] to evaluate the fitness [
4] of the target protein in complex with the docked compound
[5]. While sampling and scoring being the two major components of docking, a wide range of docking platforms are available to perform fast docking, offering a wide array of protocols (and scoring functions), such as Dock6
[6], Autodock Vina
[7], Glide
[8], Surflex
[9], Cluspro [
10] and many others. The 3D atomic model (structural coordinates) of the target protein (receptor) is a pre-requisite to have for docking
per se which could be either experimentally solved or computationally modeled. For the later case (in absence of an experimental template), one needs to do intensive modeling to generate the best representative structure or set of structures
[11][12]. Such approaches are often adopted for
IDPs / IDPRs in combined docking and dynamics
in silico experiments [
13,
14]. In the past, SBDD (
Figure 1) has widely been used to mitigate the effects of non-synonymous (and/or deleterious) mutations related to many common diseases.
Examples include p53, an enigmatic multi-functional protein with intrinsically disordered regions [
15], the so-called “guardian protein” in cancer, functioning as a tumor-suppressor
[16]. Again, there are specific mutations in p53 that result in the malfunctioning of the protein and increase the risk of cancers
[17]. In cancer patients, mutations acting against p53 in its DNA binding ability (in its role of a transcription factor) are frequently observed. Rescuing (or supplementing) the native function(s) in the ‘mutant p53 protein’ is one central objective in current cancer research
[18][19]. In the past, it has been shown that the small molecules can be designed to bind to
an adaptive protein–protein interface [
20] and can be made to stabilize the DNA binding domain and rescue mutant functions
[21]. Recent computational recent in an aligned direction modeled the wild-type p53 and several of its deleterious mutants
[22] shaded light on the mechanism of p53 reactivation
[22]. A novel transiently open L1/L3 pocket was identified and indicated the exposure of Cys-124 was pivotal in the formation of cavities
[22]. It was a crucial finding since Cys-124 was suggested to be the 'covalent' docking site for known alkylating p53 stabilizers
[23]. This made interest and scope to pursue a thorough screening of potential compounds, docked onto this pocket to search for other potential stabilizers. As a matter of fact, 1,324 compounds from the NCI/DTP Open Chemical Repository Diversity Set II were docked onto the generated ensemble structures of the R273H cancer mutant, out of which 45 compounds were chosen for biological assay
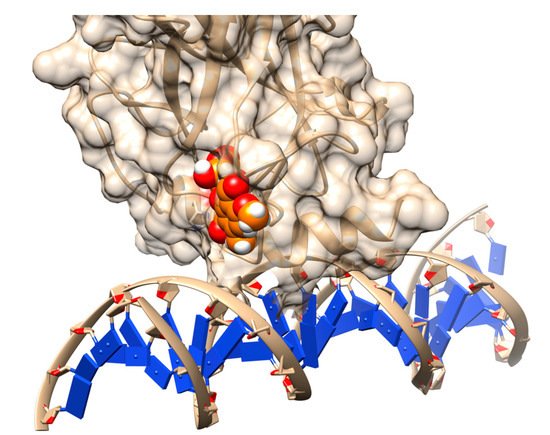
[22]. Finally, stictic acid (NSC-87511) (
Figure 2) was experimentally validated to be an efficient reactivation compound for mutant p53
[22].