Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biophysics
Protein-DNA interactions are the core of the cell’s molecular machinery. For a long time, conventional biochemical methods served as a powerful investigatory basis of protein-DNA interactions and target search mechanisms. Currently single-molecule (SM) techniques have emerged as a complementary tool for studying these interactions and have revealed plenty of previously obscured mechanistic details.
- protein-DNA interaction
- single-molecule
- DNA immobilization
- fluorescence microscopy
- nanobiotechnology
1. Introduction
Protein-DNA interactions are fundamental to the existence of all living organisms. At the cellular level, these interactions govern different biomolecular processes, such as compaction and recombination of genetic material [1,2], DNA repair [3,4] and replication [5], and transcription and its regulation [6,7]. In order to combat bacteriophage infections, bacteria and archaea employ diverse protein-based defense systems-restriction-modification [8], CRISPR-Cas [9], and others. All of them are based on the ability of proteins to specifically interact with their invading DNA target sequences.
2. Traditional DNA Curtains
2.1. Technological Design
A novel next-generation in vitro SM biophysical technique—DNA curtains—is by far the most superior type of DNA flow-stretch assay, as it is capable of imaging hundreds or even thousands of separate fluorescently labeled protein-DNA complexes at once in real time [100]. The high-throughput nature of DNA curtains determines one of its fundamental advantages over the other similar approaches—in conjunction with TIRF microscopy, this technology enables the collection of large amounts of statistical data during a single experiment [101]. As well as the other types of DNA flow-stretch assays, this experimental platform can provide a whole set of information regarding the SM dynamics of protein-DNA interactions, in particular, when relatively long DNA substrates are employed. Real-time visualization of protein translocation along the DNA molecule, complete characterization of DNA-binding landscapes, and the assessment of protein dwell times—all of this can be efficiently accomplished by the means of DNA curtains [102].
The technological principle of conventional DNA curtains is implemented through the combination of supported lipid bilayers (SLBs) and lipid diffusion barriers (Figure 2A) [100]. In this approach, the surface of a flow cell is initially passivated with an SLB, which has a fraction of its lipid heads functionalized with biotin. Afterwards, this lipid bilayer is coated either with neutravidin or sAv and the biotinylated DNA end is then immobilized on this protein monolayer. The fluidity of the lipid bilayer ensures the long-range 2D motion of lipid-tethered DNA. By applying a hydrodynamic flow, such DNA molecules can be organized at the lipid diffusion barriers, which are deployed on the flow cell surface and are perpendicular to the buffer flow direction (Figure 2B). These physical barriers disrupt the lateral diffusion of the lipids, as they cannot physically cross such barriers, and that results in the accumulation of lipid-tied DNA molecules along the leading edges of the barriers. Being under the continuous hydrodynamic force, hundreds or even thousands of individual DNA molecules not only align in a parallel manner along these diffusion barriers, but they also stretch along the surface of the flow cell. Such a strategy allows one to visualize these DNA molecules with TIRF microscopy (Figure 2C,D) [99,101].
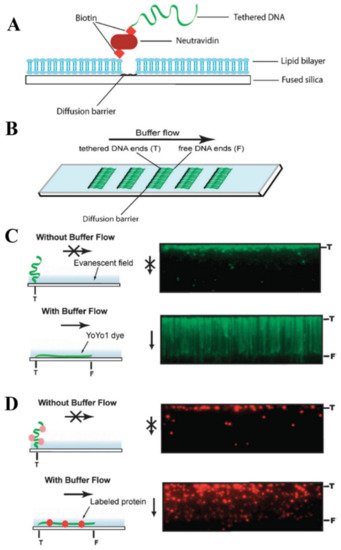
Figure 2. Technological design of high-throughput traditional DNA curtains. (A) Side view schematically representing the fundamental components that form conventional DNA curtains. (B) Top view of a microscope slide with several arrays of DNA curtains on its surface. (C) Left: schematic representation of surface-tethered DNA molecule response to the buffer flow changes. Right: total internal reflection fluorescence (TIRF) microscopy images showing the response of individual DNA molecules organized in a parallel manner to the application and stopping of a hydrodynamic flow. In the absence of a buffer flow, DNA molecules diffuse away from the glass surface and leave the evanescent field. Here, DNA is stained with the intercalating YOYO1 fluorescent dye. (D) A representative experiment, in which traditional DNA curtains are used to directly observe single protein-DNA interactions. In this case, the DNA molecules are not stained, while the investigatory protein (chromatin remodeling enzyme Rdh54) is fluorescently labeled using a quantum dot. Stopping of the hydrodynamic flow results in the out of evanescent-field diffusion of individual DNA-bound protein molecules. Left: schematic drawing. Right: TIRF microscopy images. Reprinted from [44], with permission from Royal Society of Chemistry.
The employment of SLBs determines another substantial advantage of traditional DNA curtains. Lipid bilayers are splendidly compatible with SM biochemical experiments since in the flow cell they mimic a similar microenvironment that is typically found inside the cell. Lipid bilayers can be readily formed, and they also tolerate a variety of solution conditions [50]. In addition, lipid bilayers can be easily modified by incorporating synthetic lipids with substituted heads, whereas bilayers composed of zwitterionic lipids help to avoid non-specific surface-adsorption of DNA and distinct proteins in the range of near-neutral pH values.
Another essential component of conventional DNA curtains’ technological design—lipid diffusion barriers—can be fabricated by several techniques. The most straightforward approach is the manual etching of diffusion barriers, when the surface of a glass slide is engraved with a diamond-tipped drill [50]. This method does not require any specialized equipment, yet it does not allow control of the width, depth, and arrangement of the etched barriers. For this reason, diffusion barriers are usually formed by the means of nanofabrication which enables surface patterning of a nanometer precision [103]. In electron beam lithography, a microscope slide is at first covered with a thin polymeric film—polymethylmethacrylate (PMMA) bilayer—on top of which a layer of an antistatic agent (Aquasave) is then deployed. Next, the electron beam is used to etch the desired pattern in the polymeric film, thus exposing the underlying glass surface. The metal (typically chromium) is then evaporated in vacuum and deposited on the entire surface including PMMA and the uncovered glass. The remaining polymer is peeled off during the lift-off procedure, as only the metallic structures remain on the microscope slide. These metal assemblies, ultimately, serve as robust lipid diffusion barriers that do not interfere with the fluorescence imaging of the immobilized DNA molecules [100,104,105]. Fabrication of physical barriers can also be implemented through the nanoimprint lithography which is rather similar to the one described above, as this technique conjoins the utilization of electron beam and lift-off with the inductively coupled plasma etching [106]. Although electron beam and nanoimprint lithographies permit the high-resolution patterning of glass substrates, they are restricted by their low-throughput nature, as both these lithographic techniques require the raster scanning of an electron beam along each nanobarrier segment, which limits the overall number of barriers formed on the surface of each slide. Another drawback is the necessity of high-cost specialized equipment. To overcome these shortcomings, a rapid, cost-effective, UV-lithography-based method has been recently developed, enabling the high-throughput manufacturing of chromium-based physical barriers [107].
Lipid diffusion barriers can also be fabricated out of a whole spectrum of different materials. During the production of these nanobarriers by electron beam or nanoimprint lithography, vapor deposition of thin films, composed of such metals or oxides as chromium, titanium oxide, or gold, is usually performed on the surface of a glass slide [50,100,103,108]. Physical barriers impeding the lateral diffusion of lipids can also be made from hydrogen silsesquioxane [104]. This negative resistance eliminates the need for metal evaporation, thus simplifying and shortening the whole process of nanobarrier fabrication. Yet, most of the time, lipid diffusion barriers are formed out of chromium. Since this approach is fairly expensive and time-consuming, chromium-patterned microscope slides are usually reused, which means that such slides must be thoroughly cleaned after each experiment in order to avert the interference of surface-remaining organic matter with the subsequent experiments. However, strong cleaning agents (acetone, acids) and harsh cleaning methods (sonication, boiling) tend to damage chromium barriers; with mild cleaning materials, these barriers still wear out after several reuse cycles, and they also begin to scatter the excitation beam. Recently, it was demonstrated that DNA curtains can be assembled using nanotrenches as barriers to the lateral diffusion of lipids [105]. Nanofabrication of trenches is achieved by the means of electron beam lithography and reactive ion etching, where these physical barriers of different nature are engraved on the glass slide surface. Nanotrenches are robust and do not degrade even under extremely harsh cleaning conditions and that enables the complete removal of any residual surface contaminants. This, in turn, ensures the reliable and repeated reuse of patterned microscope slides and allows one to assemble well-organized high-quality DNA curtains during each successive experiment.
2.2. Single-Tethered DNA Curtains
To address the need for specific conditions dictated by a broad diversity of SM experiments, DNA curtains can be prepared in several different configurations. The most common of them is single-tethered DNA curtains, in which individual DNA molecules are immobilized on a lipid bilayer by one of their ends [109]. TIRF imaging of such DNA molecules is only possible under the conditions of a continuous buffer flow that stretches the DNA along the glass surface. The advantage of this configuration is the ability to temporarily stop the fluid flow, as during the experiment this can be utilized as a standard control to verify if the observed solitary DNA and protein molecules are not non-specifically bound to the flow cell surface. In addition, the constant buffer flow permits one to maintain the same tension for all anchored DNA molecules. Single-tethered DNA curtains can be assembled by employing either linear or zigzag barriers to lipid diffusion (Figure 3A,B) [100,110]. Linear barriers are rather simple, but they can lead to the overlapping of individual surface-tethered DNA molecules, which is undesirable in certain types of experiments. This is typically avoided with the use of zigzag barriers that ensure the spatial separation of each DNA molecule from its proximate neighbor.
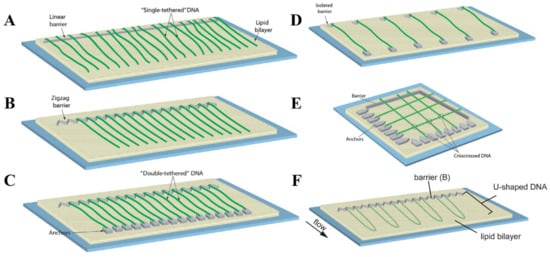
Figure 3. Configurational diversity of conventional DNA curtains. (A) Single-tethered DNA curtains assembled using a linear barrier to lipid diffusion. (B) Single-tethered DNA curtains assembled utilizing a zigzag barrier to lipid diffusion. The distance between neighboring DNA molecules is determined by a zigzag pattern. (C) Double-tethered DNA curtains. In this configuration, one end of an individual DNA molecule is immobilized on a supported lipid bilayer, while the other DNA end is typically tethered to a downstream pedestal via a specific digoxigenin–anti-digoxigenin interaction. (D) DNA curtains assembled employing a parallel array of double-tethered isolated (PARDI) molecule patterns. Such a configuration of DNA curtains grants the possibility to maintain an extremely low local concentration of surface-immobilized DNA molecules during the experiment. (E) Crisscrossed DNA curtains. Here, the intersections (marked with circles) between the crisscrossed DNA molecules represent the regions of locally high DNA concentration. (F) U-shaped DNA curtains. Since individual DNA molecules in this configuration are anchored on a lipid bilayer by both of their ends, application of a hydrodynamic flow forces the DNA molecules to adopt a “U” shape during their alignment at the lipid diffusion barriers. (A–E) are adapted from [102], with permission from Elsevier. (F) is adapted from open-access source [108].
Single-tethered DNA curtains were utilized for probing the SM dynamics of a broad spectrum of DNA-binding proteins in real time. Abo1 is one of those proteins for which biological functions have been recently uncovered by the use of DNA curtains assembled in a single configuration [111]. Single-tethered DNA curtains were also combined with biochemical assays to gather more molecular details about Abo1-mediated histone loading onto DNA and the ongoing interactions between Abo1 and DNA [112]. Another study combined single-tethered and double-tethered DNA curtains to monitor the motion of individual fluorescently labeled Type I-E CRISPR-Cas proteins on DNA [113]. It revealed that Cas1-Cas2 integrase is able to non-specifically sample the DNA through 3D collisions with a short lifetime, while Cas3 helicase/nuclease translocates on DNA by using two main modes, as at the target sequence it either moves in conjunction with the 11-subunit crRNA-containing Cascade surveillance complex or independently away from Cascade. In the other study, the employment of single-tethered DNA curtains and catalytically dead RNA-programmable DNA endonuclease CRISPR-Cas12a enabled the SM-level real-time fluorescence imaging of artificial virus-like nucleocapsids self-assembly kinetics [114]. Furthermore, single-tethered DNA curtains were applied for directly probing the behavior of individual quantum-dot-labeled DNA translocating proteins [115,116,117]. In these studies, to probe the crowding effect on protein-of-interest translocation, single-tethered DNA curtains were assembled using DNA substrates that were occupied by different proteins, such as EcoRIE111Q, E. coli RNA polymerase holoenzyme, lac repressor, or nucleosomes. Single-tethered DNA curtains were also utilized in the SM-level mechanistic studies of diverse proteins participating in the cellular processes of DNA end resection, such as human resectosome [118], mycobacterial heterodimeric helicase-nuclease AdnAB [119,120], DNA-dependent protein kinase [121], poly(ADP-ribose) polymerase-1 [122], and exonuclease 1 [123]. Moreover, this type of next-generation in vitro DNA flow-stretch assay was proven to be suitable for the efficient studies of discrete interactions between the DNA and Rad51 recombinase, the key component of the eukaryotic HR machinery [124,125,126,127,128,129].
2.3. Double-Tethered DNA Curtains
Certain SM experiments are better performed without a continuous buffer flow, as some of them involve the usage of expensive reagents, while others cover the investigation of individual protein interactions with the surface-immobilized DNA molecules. It is in the latter case that the hydrodynamic force induced by the flow of a buffer can potentially affect the behavior of proteins or their complexes, leading to the disruption of protein-DNA interactions or causing some undesired bias, for instance, during the target search or when a protein is travelling along the DNA. In double-tethered DNA curtains, discrete DNA molecules are immobilized on the surface by both of their extremities, enabling the visualization of the entire DNA contour length in the absence of any fluid flow (Figure 3C) [130]. This configuration is implemented through the use of two structural elements: linear or zigzag barriers and pedestals that are located downstream of these obstructions to lipid diffusion. Single end of the DNA molecule is initially tethered to the lipid bilayer through a specific biotin-sAv interaction. By applying a buffer flow, such DNA molecules are then aligned in a parallel manner at the physical barriers and stretched along the glass surface. Since the other end of the extended DNA molecule is modified with digoxigenin, it is lastly attached to the downstream pedestals coated with digoxigenin antibodies. Therefore, double configuration of DNA curtains ensures that all of the separate surface-fixed DNA molecules have an identical orientation with respect to their nucleotide sequence.
As well as single-tethered ones, DNA curtains assembled in a double configuration were extensively applied in the multitude of mechanistic studies of individual protein-DNA interactions. Single- and double-tethered DNA curtains in tandem with ensemble biochemical measurements were employed to investigate the target search mechanisms of S. pyogenes RNA-programmable DNA endonuclease Cas9 belonging to Type II CRISPR-Cas systems [131]. By utilizing double-tethered DNA curtains, real-time SM fluorescence imaging of target search of E. coli Cascade nucleoprotein complex on λ-DNA was performed, which showed that such a process can be implemented in two distinct pathways—PAM-dependent or PAM-independent [132]. This kind of next-generation in vitro DNA flow-stretch assay was also employed for SM characterization target search mechanisms of various proteins including: Type I-E CRISPR-Cas interference and primed acquisition complexes [133], hexameric DNA translocase FtsK without obstacles on DNA [134], and in the presence of obstacles [135]. Furthermore, DNA curtains assembled in a double configuration were used to observe rapid, highly processive, and ATP-hydrolysis-dependent translocation of S. cerevisiae quantum-dot-labeled condensin along DNA at the SM level [136]. This SM biophysical technique was also utilized to probe the movement of Mlh1-Pms1 along DNA by which this complex locates the lesion-bound Msh2-Msh6 proteins during the eukaryotic postreplicative mismatch repair [137]. Similarly, double-tethered DNA curtains enabled the disclosure of lesion-search mechanism characteristic of Msh2-Msh3 proteins, as it was shown that this eukaryotic mismatch repair complex uses 1D sliding and microscopic hopping to scan individual DNA molecules, which determines its ability to bypass nucleosomes and other protein roadblocks during the lookout for DNA damage [138]. There are several SM-level studies of proteins involved in HR which also make use of DNA curtains assembled in either single or double configuration [139,140,141,142].
2.4. Other Configurations
The configurations described above are the two fundamental types of DNA curtains, as they are typically employed for the efficient mechanistic studies of protein-DNA interactions at the SM level. Still, there are some other occasionally used configurations of traditional DNA curtains which are designed for more specialized SM experiments. DNA curtains can be assembled using parallel array of double-tethered isolated (PARDI) molecule patterns (Figure 3D) [143]. This configuration ensures a 7 µm or even larger spacing between adjacent double-tethered DNA molecules, which means that during the experiment the local concentration of DNA is maintained at an extremely low value. Thus, DNA curtains based on PARDI patterns are suitable for studying individual protein-DNA interactions, in particular protein association kinetics, under the conditions where the effects of locally high DNA concentration are absent. It is the complete opposite with crisscrossed DNA curtains, as in this kind of configuration, high local DNA concentration is established by perpendicularly crossing solitary double-tethered DNA molecules (Figure 3E) [144]. Such DNA molecules are separated one from another at an average of ~106 nm and the intersections between them are considered to be locally high DNA concentration regions, which can be utilized for investigating the capabilities of different proteins to travel between two DNA substrates located in close proximity to each other. There are also U-shaped DNA curtains, in which both biotinylated ends of the individual DNA molecules are initially immobilized on a lipid bilayer (Figure 3F) [108]. The continuous buffer flow is then applied, causing the DNA to adopt a “U” shape, as double-tethered DNA molecules are aligned at the zigzag barriers to lipid diffusion. Such a configuration is especially beneficial for studying distinct mechanisms of DNA compaction, such as loop extrusion induced by condensins, at the SM level, since it enables real-time visualization and tracking of DNA-length and conformational changes during the individual DNA looping events.
2.5. Single-Stranded DNA Curtains
dsDNA is not the only DNA substrate that can be utilized for the assembly of conventional DNA curtains. In most biochemical reactions that comprise the processes of DNA repair and replication, ssDNA plays a key role as an intermediary product. To achieve a deeper understanding of these biomolecular processes, the experimental tool of ssDNA curtains was developed, enabling the fluorescence imaging of surface-tethered stretched ssDNA molecules at the SM level [145,146,147,148]. During the assembly of ssDNA curtains, the synthesis of long ssDNA molecules is at first performed by the means of a rolling circle replication, as φ29 DNA polymerase, a circular ssDNA template and a biotinylated DNA primer are utilized for this reaction (Figure 4A) [149]. The ssDNA products of a rolling circle replication are immobilized on the lipid bilayer through biotin-sAv interaction, and by applying a buffer flow, they are then aligned in a parallel manner along the leading edges of the diffusion barriers. However, optical imaging of such ssDNA molecules is hampered by high ssDNA compactness and the inability to fluorescently label it with intercalating dyes, as they compromise the integrity of the ssDNA molecules. To overcome these limitations, replication protein A (RPA) fused with enhanced green fluorescent protein or mCherry protein is usually employed, as RPA is capable of binding to the ssDNA and removing its secondary structure [146,147]. RPA-ssDNA filaments are much stiffer than the uncovered ssDNA and that allows RPA-bound ssDNA molecules to be stretched under a constant buffer flow and visualized by the means of TIRF microscopy. ssDNA curtains can also be assembled in a double configuration, in which RPA-ssDNA complexes are non-specifically adsorbed on the exposed anchor points located downstream of the lipid diffusion barriers (Figure 4B) [145,147]. Another extension of ssDNA substrate-based DNA curtains is low-complexity ssDNA curtains that allow one to simultaneously synthesize and visualize individual low-complexity ssDNA molecules at the SM level in real time [150]. These DNA curtains are assembled using a phosphorylated template, which is designed in such a manner that standard Watson–Crick base pairing in the generated ssDNA product is non-existent, a biotinylated primer, T4 DNA ligase, and φ29 DNA polymerase. After a primer is annealed with the template, T4 DNA ligase is employed to produce closed minicircles that are then injected into the flow cell. During a rolling circle replication with φ29 DNA polymerase, ssDNA molecules in length of >50,000 nucleotides are synthesized, and then organized at lipid diffusion barriers by employing a buffer flow. Since the produced ssDNA consists of only thymidine and cytidine, the number of possible secondary structures in ssDNA is minimized and the stretching of this molecule can be achieved by applying only minimal force. Such a unique feature of low-complexity DNA curtains makes this kind of experimental platform especially beneficial for investigating the ssDNA physical changes induced by its interactions with other nucleic acids or ssDNA-binding proteins.
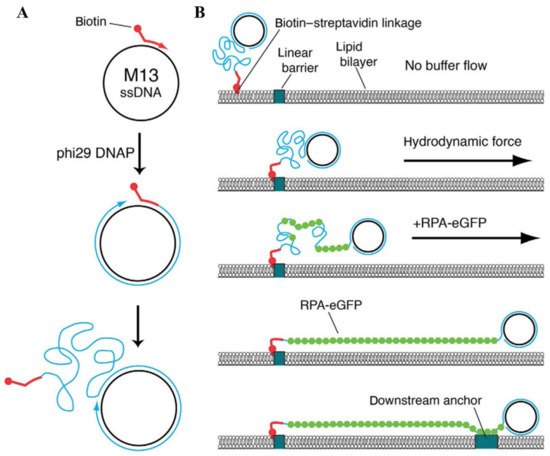
Figure 4. Assembly of ssDNA curtains. (A) Biotinylated ssDNA substrate is produced by a rolling circle replication. A biotinylated primer, circular ssDNA template such as M13mp18, and φ29 DNA polymerase are employed for this kind of DNA replication. (B) During the assembly of ssDNA curtains, a biotinylated ssDNA substrate is initially immobilized on a supported lipid bilayer and aligned at the lipid diffusion barriers by applying a hydrodynamic flow. Next, RPA-eGFP is introduced into the flow cell. This fusion protein fluorescently labels the ssDNA molecule and removes its secondary structure. As RPA-eGFP enters the channel of a flow cell, ssDNA becomes visible, as it gradually unwinds and stretches along the surface. To assemble double-tethered ssDNA curtains, RPA-ssDNA complex is non-specifically adsorbed on the exposed pedestals deployed downstream of the lipid diffusion barriers. Reprinted from [149], with permission from Elsevier.
ssDNA curtains are most suitable for investigating ssDNA intermediates involving cellular processes, such as DNA replication and HR, at the level of a single molecule. This experimental tool enabled high-throughput SM fluorescent imaging of RADX, a mammalian ssDNA-binding protein that preserves uncapped telomeres and stalled replication forks, and interactions with ssDNA substrates [151]. Another study combined ssDNA curtains with bulk biochemical assays to examine the molecular mechanisms by which RADX regulates RAD51 activity and DNA replication [152]. ssDNA curtains were utilized to probe the mechanism employed by RAD51 paralogs to remodel and stabilize RAD51 filaments [153], investigate the homology search—alignment and pairing of ssDNA with a homologous duplex DNA—of S. cerevisiae RAD51 [154], examine the interplay between S. cerevisiae RPA, RAD52, and RAD51 during the assembly of a presynaptic complex [155], monitor the interactions between human RPA and human RAD51 during presynaptic complex formation [156], and study the properties of presynaptic complexes composed of two S. cerevisiae DNA recombinases—RAD51 and DMC1, of which the latter is a meiosis-specific enzyme [157]. ssDNA curtains in conjunction with the biolayer interferometry measurements allowed one to investigate the behavior of human RECQ5 helicase, which exhibits anti-recombinase activity by negatively regulating RAD51 during HR, loaded on different nucleoprotein complexes at the SM level in real time [158]. Moreover, real-time visualization of individual S. cerevisiae Srs2 superfamily 1 helicase, which is another anti-recombinase maintaining genome integrity through the disassembly of toxic recombination intermediates, complexes acting upon long RAD51-bound ssDNA substrates, was performed using ssDNA curtains [159]. In combination with additional methods, ssDNA curtains were likewise applied in SM-level mechanistic studies of other HR-related proteins, such as RAD52 [160,161], RAD54 [162,163], and RPA [164,165,166].
2.6. Combination with Other Techniques
There are also a few successful examples of traditional DNA curtains being coupled with other manufacturing and biophysical techniques. In order to fully characterize a biochemical reaction, it is commonly necessary to perform multiple long-term experiments of the same nature, in which different reaction conditions related to the changes in protein composition or nucleotide or salt concentration can be thoroughly tested. For this reason, DNA curtains platform was combined with polydimethylsiloxane (PDMS)-based technology of microfluidics [167]. This allowed one to enhance the throughput of DNA curtains tool even further, providing an opportunity to concurrently investigate up to five distinct biochemical reaction conditions at the SM level. Another example is the TIRF-trap microscope—a combination of DNA curtains with OTs—that was developed for investigating the physical properties of separate DNA molecules organized in single-tethered DNA curtains configuration [168]. In this setup, the biotinylated end of a DNA molecule aligned at a nanofabricated diffusion barrier is immobilized on an sAv-covered lipid bilayer, while the other DNA extremity is attached to a fluorescently labeled laser-trapped bead via a digoxigenin–anti-dig linkage. Therefore, this experimental tool not only permits the high-throughput imaging of individual protein-DNA interactions in real time, but it also allows one to simultaneously perform force-based measurements, as the response of these biomolecular complexes to the applied external force can be observed directly.
3. Other Types of DNA Curtains
The technological design of conventional DNA curtains is implemented solely through the use of two fundamental components—SLBs and nanobarriers to the lateral diffusion of lipids. Nevertheless, they likewise confer several shortcomings on this next-generation SM approach. Preparation of lipid-bilayer-based systems may pose some experimental risks related to system stability and defect management. Meanwhile, nanofabrication of physical barriers, which can be executed by the diverse production methods discussed above, is time-consuming, technically challenging, and requires not only specific knowledge in the corresponding field of manufacturing, but also expensive equipment with limited availability. These limiting factors create the need for the development of innovative strategies for assembling DNA curtains in such a way that all the experimental capabilities offered by this platform would be completely retained, whilst banishing the aforementioned drawbacks.
Suspended DNA curtains are somewhat capable of fulfilling such requirements, as this type of DNA curtain provides a few significant advantages over the traditional ones, although it falls short of some basic features. In suspended DNA curtains, individual DNA molecules are, at first, attached to a gold nanowire, which bisects the microfluidic channel of a flow cell, via biotin-sAv interactions, and are then organized in a parallel manner by a buffer-flow-induced stretching [169]. Since this nanowire is elevated, the DNA molecules coupled to it are suspended further away from the flow cell surface, which prevents the DNA molecules from non-specifically interacting with the surface and should, in theory, allow one to only observe those proteins that are bound to DNA. This kind of configuration also determines the overall lower buffer flow rates necessary to achieve the maximum extension of nanowire-tethered DNA molecules, as they experience a more rapid and uniform hydrodynamic flow due to the elevation. However, suspended DNA curtains are still hardly applicable for efficient studies of protein-DNA interactions since the assembling strategy, which this SM platform is based on, results in substantial overlapping of individual nanowire-anchored DNA molecules and, most importantly, it compromises the fundamental feature of DNA curtains—the high-throughput imaging capabilities offered by this technology.
Another type of these next-generation in vitro DNA flow-stretch assays that perfectly meet the criteria stated above is soft DNA curtains [170]. In this recently developed version of DNA curtains, corresponding scanning probe microscopy and soft lithography techniques—AFM and protein lift-off microcontact printing—are combined together, enabling the precise nanopatterning of chemically modified glass coverslip surfaces with protein line-features (Figure 5A). At first, the flat side of PDMS stamp is covered with sAv or traptavidin (tAv) by inking and then the protein ink drop is sucked with the pipette, followed by the washing and drying of the elastomeric stamp. Next, inked PDMS stamp is placed on the Si master, surface of which has inscribed nanometer-sized lines. At this stage, a controllable printing pressure acts on the formed PDMS-master “sandwich”, as this force is generated using a home-built portable printing device. The stamp is then removed from the Si master, determining the selective subtraction of certain protein regions covering the surface of the stamp, and is transferred onto the glass coverslip, the surface of which is coated with a layer of methoxy-PEG and biotin-PEG molecules. At this stage, a controllable printing pressure generated by the home-built portable printing device also acts on the formed PDMS-coverslip “sandwich”. Finally, the stamp is removed from the chemically modified glass coverslip, resulting in the formation of protein line-features on the glass surface.
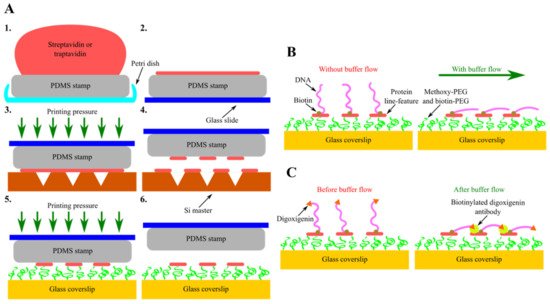
Figure 5. Assembly and configurational variety of soft DNA curtains. (A) Principal steps of soft DNA curtains assembly—inking of the elastomeric stamp (1, 2) and selective subtraction of proteins by lift-off microcontact printing (3–6). (B) Single-tethered soft DNA curtains. Individual DNA molecules biotinylated only at a single end are immobilized on streptavidin (sAv) or traptavidin (tAv) line-features patterned on the glass coverslip surface. In the absence of a buffer flow, the free end of such DNA molecules is diffused further away from the surface. Upon the application of a hydrodynamic flow, these single-tethered DNA molecules stretch along the glass surface and align in a parallel manner with respect to each other. (C) Double-tethered soft DNA curtains. To assemble double-tethered soft DNA curtains, biotin and digoxigenin-functionalized DNA substrates and biotinylated anti-dig antibodies are employed. These antibodies are initially introduced into the flow cell containing the nanopatterned surface and single-tethered DNA molecules. In the presence of a buffer flow, the digoxigenin end of the stretched DNA molecules then binds to the adjacent anti-dig-coated sAv or tAv line-feature, thus ensuring that such DNA molecules stay aligned in a parallel manner and remain in an extended conformation even when the hydrodynamic flow is halted.
sAv or tAv line-features formed on the PEGylated glass coverslip surface serve as the stable anchor points for the surface immobilization of biotinylated DNA molecules. Moreover, such protein features ensure predefined alignment of the DNA molecules after immobilization. Upon the application of a buffer flow, anchored DNA molecules are stretched along the glass surface, allowing them to be visualized with TIRF microscopy (Figure 5B). Soft DNA curtains can also be assembled in a double configuration, where the digoxigenin-labeled end of the surface-tethered DNA molecule is attached to the adjacent protein line-feature covered in biotinylated digoxigenin antibodies by the means of a hydrodynamic flow (Figure 5C) [171]. Same as the traditional ones, soft DNA curtains enable the high-throughput fluorescence microscopy imaging of individual DNA molecules organized in a massively parallel manner, whereas double-tethered soft DNA curtains also ensure the defined orientation of surface-immobilized DNA. For instance, soft DNA curtains were employed to observe the restriction endonuclease BfiI-catalyzed hydrolysis of discrete λ-DNA molecules and to visualize the dynamics of S. pyogenes Cas9 specific interactions with the double-tethered λ-DNA. Nevertheless, the eliminated need for expensive, specific-handling-knowledge-requiring equipment and the ingenious technological design of soft DNA curtains render them a cost-effective, simple, versatile, and user-friendly platform for efficient SM-level studies of protein-DNA interactions that is worth having in one’s scientific toolbox even for users with a little experience in this research field.
Moreover, an alternative to the whole technology of DNA curtains—DNA skybridge—has been developed recently [172]. As well as DNA curtains, DNA skybridge enables the high-throughput imaging of protein-DNA interactions at the SM level. However, by utilizing some unique strategies, this novel tool fulfills the key concept of DNA curtains somewhat differently. A special 3D structure consisting of 4 µm high thin quartz barriers serves as a basis for the assembly of DNA skybridge. One biotinylated end of an individual DNA molecule is initially immobilized on the sAv-coated apex of the barrier. Upon the application of a buffer flow, this DNA molecule is then stretched, and its other end is tethered to the adjacent quartz ridge via the same biotin-sAv interaction. Ultimately, such DNA molecules are visualized using thin light sheet fluorescence microscopy. These two fundamental differences between DNA curtains and DNA skybridge permit some considerable advantages to the latter platform. The employment of the different optical imaging setup allows one to obtain high signal-to-noise ratios by reducing the background noise caused by the surface-bound fluorophores. Meanwhile, the organization of individual DNA molecules in a manner in which they are located far away from the surface eliminates the impact of false signals coming from non-specifically adsorbed fluorescently labeled proteins on the real-time SM-level visualization of dynamic protein-DNA interactions.
This entry is adapted from the peer-reviewed paper 10.3390/applnano3010002
This entry is offline, you can click here to edit this entry!