Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
Interleukin (IL)-18 was originally discovered as a factor that enhanced IFN-γ production from anti-CD3-stimulated Th1 cells, especially in the presence of IL-12. Upon stimulation with Ag plus IL-12, naïve T cells develop into IL-18 receptor (IL-18R) expressing Th1 cells, which increase IFN-γ production in response to IL-18 stimulation.
- IFN-γ
- Inflammation
1. Introduction
Th1 cells produce interferon (IFN)-γ upon stimulation with antigen (Ag) plus antigen presenting cells or anti-CD3 antibody in vitro and in vivo. Lipopolysaccharide (LPS)-stimulation of anti-CD3-stimulated Th1 cells does not induce the production of IFN-γ in vitro. However, the injection of LPS into Propionibacterium acnes-primed mice or Bacillus Calmette–Guerin (BCG)-infected mice, but not naïve mice, strongly induced IFN-γ production in vivo [1][2]. Furthermore, to our surprise, the addition of sera derived from P. acnes-primed and LPS-challenged mice strongly enhanced IFN-γ production by anti-CD3-stimulated Th1 cells in vitro, suggesting the presence of IFN-γ inducing factor(s) in the sera.
Because IL-12 is produced by LPS-stimulated macrophages and dendritic cells (DC), IL-12 from LPS-stimulated macrophages or DC in P. acnes-primed mice were initially thought to induce anti-CD3-stimulated Th1 cells to produce IFN-γ in vivo and in vitro. Indeed, the sera contained high levels of IL-12. However, only the addition of sera from P. acnes-primed and LPS-challenged mice, but not the addition of excess doses of IL-12, enhanced the production of IFN-γ from anti-CD3-stimulated Th1 cells, strongly suggesting the presence of IFN-γ inducing factors in the sera from P. acnes-primed and LPS-challenged mice.
Physicochemical studies and amino acid sequence analysis revealed that IFN-γ inducing factor (IGIF) is different from IL-12. The molecular cloning of IGIF was performed by Okamura in collaboration with Hayashibara Biochemical Laboratories. Soon after human IGIF was cloned [3], we and others found various functions of IGIF, including the induction of IL-2 production, IL-2 receptor (IL-2R) and Fas ligand (FasL) expression on Th1 cells, and the activation of natural killer (NK) cells. Based on these pleiotropic functions of IGIF, we named IGIF “IL-18” [2]. Although both IL-12 and IL-18 are major factors in IFN-γ production by Th1 cells, IL-12 is a differentiation factor that induces the development of Th1 cells—in contrast, IL-18 is a proinflammatory cytokine that facilitates IFN-γ production by Th1 cells particularly in conjunction with IL-12. Indeed, IL-12 and IL-18 from LPS-stimulated macrophages synergistically induced IFN-γ production from Th1 cells in P. acnes-primed and LPS-challenged mice.
Murine and human IL-18 proteins consist of 192 and 193 amino acids, respectively [1][3]. Based on the homology of its amino acid sequence compared with IL-1β, IL-18 is classified as a member of the IL-1 cytokine family. Human IL-18 and IL-1β share only 15% sequence homology although they contain a common β-pleated sheet structure. Furthermore, similar to IL-1β, IL-18 is produced as a biologically inactive precursor, pro-IL-18, which lacks a signal peptide and requires proteolytic processing to become active. The cleavage of pro-IL-18 or pro-IL-1β depends mainly on the action of the intracellular cysteine protease caspase-1 in the NLRP3 inflammasome [4][5].
The IL-18 receptor (IL-18R) consists of the inducible component IL-18Rα (IL-1 receptor-related protein [IL-1Rrp]) and the constitutively expressed component IL-18Rβ (IL-1R accessory protein-like [IL-1RAcPL]) [2]. Cytoplasmic domains of IL-18Rα and IL-18Rβ contain a common domain termed the Toll-like receptor (TLR)/IL-1R (TIR) domain, shared by other IL-1R family members and TLRs. Upon stimulation with IL-18, IL-18Rα forms a high-affinity heterodimeric complex with IL-18Rβ—which mediates intracellular signal transduction. Cytoplasmic TIR domains of the receptor complex interact with myeloid differentiation primary response 88 (MyD88), a signal adaptor containing a TIR domain [6], via TIR-TIR interactions. Then, MyD88-induced events result in the activation of nuclear factor (NF)-κB and mitogen-activated protein kinase (MAPK) via association with the signal adaptors IL-1R-associated kinase (IRAK) 1-4 and tumor necrosis factor (TNF) receptor-activated factor (TRAF) 6, respectively, which eventually leads to the appropriate gene expressions, such as Ifng, Tnfa, Cd40l, and FasL.
Although IL-18 was originally discovered as a factor that induces IFN-γ production from Th1 cells, it also acts on non-polarized T cells, NK cells, NKT cells, B cells, DC and macrophages to produce IFN-γ in the presence of IL-12. Moreover, IL-18 without IL-12 but with IL-2 induces Th2 cytokine production from CD4+ NKT cells, NK cells, and even established Th1 cells. Furthermore, IL-18 with IL-3 induces mast cells and basophils to produce IL-4 and IL-13. Therefore, IL-18 stimulates both innate immunity and acquired immunity [2][7].
The source of IL-18 was initially demonstrated to be from Kupffer cells, which constitutively express pro-IL-18. In addition, LPS binding to TLR4 induces the production of IL-18 via the activation of caspase-1. In contrast, upon stimulation with LPS, DC or macrophages increase their transcription of pro-IL-18 mRNA and subsequently their production of pro-IL-18, which is then processed by caspase-1 to be secreted as mature IL-18. In addition to these IL-18 producing cells, pro-IL-18 is produced by a wide variety of other cells, including keratinocytes, intestinal epithelial cells, and osteoblasts suggesting it has an important pathophysiological role in health and disease. Like other cytokines, IL-18 shows its pleiotropic action depending on its cytokine milieu (Figure 1).
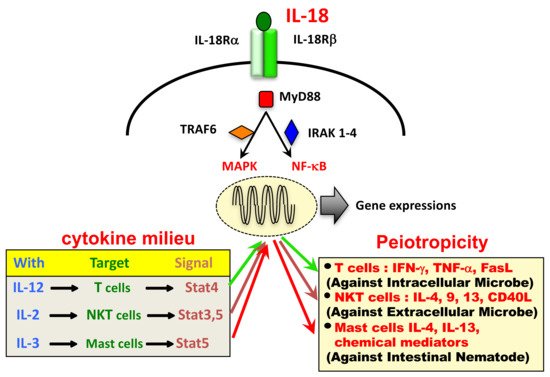
Figure 1. Pleiotropic action of IL-18 depends on its cytokine milieu.
2. IL-18 in Disease
2.1. Endotoxin-Induced Systemic and Tissue Diseases
2.1.1. Induction of Endotoxin Shock in P. acnes-Primed Mice
Sepsis is still a common, life-threatening disorder, in which endotoxin is a key player. Paradoxically, patients with high serum levels of endotoxin do not necessarily develop lethal shock, whereas some patients die of septic shock even when their serum endotoxin levels are low. To understand this paradox, we measured serum IL-6 levels of patients, because LPS induces IL-6 production in vivo. The simultaneous measurement of serum levels of LPS and IL-6 indicated that there were at least two groups: the high IL-6 group was endotoxin shock susceptible and the low IL-6 group was endotoxin shock resistant [8]. These results suggested that limiting factors determine the sensitivity of patients to endotoxin shock.
Rodents are genetically resistant to LPS. Therefore, naïve BALB/c mice are resistant to challenge with high doses of LPS (100 μg/mouse). However, BALB/c mice primed with heat-killed P. acnes, a Gram-positive skin habituating bacterium, or BCG, become highly susceptible to the lethal shock-inducing effect of LPS. Furthermore, upon LPS (1 μg/mouse) challenge, they rapidly produced IL-1, TNF-α and IL-6 and died of endotoxin shock or, if they survived, they suffered from acute liver injury through apoptosis-mediated hepatocytotoxicity [8][9]. Moreover, P. acnes-primed mice became highly susceptible to the lethal shock-inducing effects of IL-1 and TNF-α, producing high levels of IL-6 and dying after challenge with IL-1 and TNF-α. Therefore, priming with P. acnes or BCG induced lethal endotoxin shock in mice highly susceptible to LPS by the enhanced production of IL-1, TNF-α, and IL-6.
After publishing these results [8], we observed that P. acnes-primed BALB/c nu/nu mice were resistant to LPS-induced lethal shock, but died of fulminant hepatitis [1]. However, nu/nu mice reconstituted with splenic T cells died of lethal shock before the development of fulminant hepatitis after sequential treatment with P. acnes and LPS [10]. Therefore, P. acnes pretreatment rendered mice highly susceptible to the lethal shock-inducing effect of LPS by the induction of Th1 cells. Indeed, IL-12p40-deficient mice or IFN-γ-deficient mice were highly resistant to P. acnes-primed and LPS-challenged endotoxin shock, revealing the importance of IFN-γ as a limiting factor to determine the sensitivity to LPS shock.
2.1.2. LPS-Induced Liver Injury in P. acnes-Primed Mice
P. acnes-primed and LPS-challenged nu/nu mice eventually died of fulminant hepatitis. However, the administration of anti-IL-18 Ab prevented LPS-induced liver injury in P. acnes-primed nu/nu mice [1]. We found that IL-18 induced FasL expression on Th1 cells, NK cells and unique liver T cells. Therefore, IL-18 is a key player in LPS-induced liver injury and induced fulminant hepatitis through Fas-mediated hepatocytotoxicity [9]. Indeed, P. acnes-primed IL-18-deficient mice were resistant to liver injury after LPS challenge. However, the administration of IL-18 induced liver injury in P. acnes-primed IL-18-deficient mice via the induction of FasL and TNF-α [11]. Therefore, we are very interested in how IL-18 is released after LPS challenge in P. acnes-primed mice [9].
Wild type mice primed with P. acnes developed dense granulomas in the liver, and developed acute liver injury when subsequently challenged with a sublethal dose of LPS [9]. These mice had elevated serum IL-18 levels after LPS challenge. Furthermore, P. acnes-primed IL-18-deficient mice exhibited granulomas in the liver comparable with P. acnes-primed WT mice, but were resistant to acute hepatitis induced by LPS. In contrast, MyD88-deficient mice, which lack signaling common to many TLRs as well as IL-18/IL-1β signaling, primed with P. acnes had low hepatic granuloma formation and undetectable levels of IL-18 after LPS challenge [12], although MyD88-deficient Kupffer cells secreted IL-18 in response to LPS in vitro [13][9]. Therefore, we examined the contribution of TRIF for P. acnes-induced hepatic granuloma formation and LPS-induced IL-18 secretion [12]. Unlike MyD88-deficient mice, P. acnes-primed TRIF-deficient mice normally develop hepatic dense granulomas, but do not release IL-18 or develop liver injury. Therefore, we concluded that P. acnes treatment induced hepatic granuloma formation that was dependent on MyD88. Subsequent LPS challenge activated caspase-1 via the NLRP3 inflammasome and induced IL-18 release, which was dependent on TRIF, eventually leading to liver injury [12].
2.2. IL-18 in Allergy
2.2.1. Induction of IgE Production by IL-18
The daily administration of IL-18, especially with IL-2, markedly increased serum levels of IgE in naïve wild type mice [14]. An in vitro study revealed the increased expression of CD40L and production of IL-4 in CD4+NK1.1+ T cells stimulated with IL-2 and IL-18. These IL-18-stimulated NKT cells induced the development of naïve B cells into IgG1 and IgE-producing cells by the simultaneous stimulation of B cells with CD40L and IL-4 [15].
2.2.2. Innate-Type Allergic Inflammation Induced by IL-18
These mice spontaneously produced IL-18 and IgE, and developed atopic dermatitis (AD)-like skin lesions. Stat6-deficient KCasp1 Tg mice did not produce IgE, but still developed similar skin lesions. Therefore, the overproduction of IL-18 from keratinocytes induces skin lesion even in the absence of IgE [16]. We described this inflammation as “innate-type allergic inflammation” [17].
2.2.3. The Induction of IFN-γ and IL-13 Producing Super Th1 Cells by IL-2 and IL-18
Th1 cells produce both Th1 cytokines (IFN-γ) and Th2 cytokines (IL-9 and IL-13) in response to IL-18 plus IL-2. Furthermore, the intranasal administration of Ag, IL-2 and IL-18 to naïve mice bearing resting Th1 memory cells induced the development of airway inflammation and hyperresponsiveness [18]. We found that upon challenge with Ag, IL-2 and IL-18, resting memory Th1 cells produced both Th1 cytokines (IFN-γ) and Th2 cytokines (IL-9 and IL-13), which induced severe bronchial asthma. The administration of Ag and LPS also induced bronchial asthma by the induction of endogenous IL-18 from LPS-stimulated bronchial epithelial cells [19]. Therefore, Th1 cells, after stimulation with Ag and IL-18, become harmful cells that produce IFN-γ and IL-13, which induced difficult to control bronchial asthma [18][19]. We termed pathological Th1 cells as “super Th1 cells”, because they induced difficult to control asthma or AD-like skin lesions. This prominent feature of IL-18 might explain the mechanism for infection-associated allergic diseases.
2.2.4. Bronchial Asthma Induced by the Intranasal Administration of IL-2 and IL-18
The nasal administration of IL-2 and IL-18 induced airway hyperresponsiveness, pulmonary eosinophilia, and goblet cell hyperplasia in wild type mice, but not in Rag2-deficient mice [20]. However, the nasal administration of IL-33 induced similar changes in wild type mice and Rag2-deficient mice [21]. Therefore, IL-2 plus IL-18 induced pulmonary changes in a T cell-dependent manner, while IL-33 treatment induced the same changes in a T cell-independent and innate cell-dependent manner.
2.3. IL-18 in Kidney Diseases
IL-18 is well documented as being involved in various types of kidney diseases. For example, mice deficient in Il18 or those administered neutralizing anti-IL-18 Ab are resistant to acute kidney disease induced by ischemia/reperfusion [22] or by cisplatin treatment [23]. IL-18 blockade was also shown to protect against chronic kidney disease in mice induced by unilateral ureteric obstruction [24]. Recent excellent review articles have addressed this issue, in particular, focusing on its role in inflammasomes [25][26][27][28]. Here, we describe two topics of IL-18: its role in human IgA nephropathy and IL-18 as a clinical biomarker of acute kidney injury (AKI) that influences long-term outcomes of cardiac surgery.
2.3.1. Association between Serum IL-18 Levels and Renal Prognosis in IgA Nephropathy
IgA nephropathy is a primary mesangial proliferative glomerulonephritis with the prevalent deposition of IgA in mesangial cells in the glomerulus. IgA nephropathy is regarded as a benign kidney disease. However, recent clinical studies revealed that IgA nephropathy had an extremely variable clinical course and that it led to end-stage renal disease with slow progression [29][30]. It was reported that serum IL-18 levels were a potent prognostic factor for IgA nephropathy [31]. Notably, serum concentrations of IL-18 in IgA nephropathy patients were significantly elevated compared with healthy controls [31]. Patients sensitive to corticosteroid therapy showed a significant reduction in serum levels of IL-18 after therapy, while patients resistant to therapy exhibited no reduction. Moreover, the renal survival of IgA nephropathy patients with higher than median serum IL-18 levels at baseline was approximately 20% at the end of the follow-up period (four years), and approximately 80% for total IgA nephropathy patients [31]. Furthermore, immunohistochemical analyses revealed that the intensity of IL-18 and NLRP3 proteins in renal biopsy samples from patients with IgA nephropathy correlated with the severity of proteinuria [32]. Therefore, serum IL-18 concentration might be a predictor for renal prognosis in this disease.
2.3.2. Urinary IL-18 as A Biomarker of AKI after Cardiac Surgery
Because murine tubular epithelial cells secrete IL-18 and contain the components required for inflammasome activation [26][33][34], urine IL-18 levels might be elevated after acute tubular injury in human [35][36]. Urinary IL-18 is now recognized as a biomarker for AKI [37]. AKI often occurs in adults and children undergoing cardiac surgery and is a risk factor for morbidity and mortality [38][39]. Levels of serum creatinine, a biomarker for the diagnosis of AKI, increase late in the course of the disease, delaying timely treatment. Many studies have investigated new AKI biomarkers and several urinary proteins including IL-18 have been identified as early AKI biomarkers [40]. Recently, urinary biomarkers of AKI, particularly IL-18, were reported to be an additional prognostic factor for long-term postoperative mortality. Urinary IL-18 levels on post cardiac surgery days 1-3 were well correlated with the mortality rate at three-year follow-up [41]. AKI contributes to multiple organ failures [42][43], suggesting that long-term postoperative mortality might be directly evoked by AKI. However, a recent study revealed that postoperative AKI might be indicative of cardiac vascular stress, rather than an independent renal pathway for adverse cardiovascular death [44].
2.4. IL-18 in Metabolic Disorders
Early clinical studies revealed that IL-18 levels were elevated in the circulation and atherosclerotic plaques of patients with atherosclerosis [45][46]. In a prospective study of 1229 patients with coronary artery disease, at the 4-year follow-up, serum IL-18 levels were significantly higher in patients with fatal cardiovascular events than in those who did not die [47]. A community-based prospective cohort study showed that plasma IL-18 levels were a predictor of coronary evens in healthy European men [48]. Recently, a meta-analysis of the association of IL-18 with coronary heart disease identified circulating IL-18 as a possible risk factor of cardiovascular disease [49]. These reports suggest IL-18 is involved in metabolic syndrome. However, during metabolic disorders caused by excess energy, the NLRP3 inflammasome is likely to be activated by aberrant lipid metabolites and/or high glucose levels, which subsequently results in the secretion of IL-18 as well as IL-1β, which can induce inflammatory responses [50]. In contrast, IL-18 has a neutral or beneficial role in triggering obesity-associated metabolic diseases. Therefore, an increase in circulating IL-18 concentrations might be an indicator of the activation levels of the NLRP3 inflammasome in the early phase of disease. IL-18, together with IL-12 and/or IL-15, exerts proinflammatory effects such as the activation of Th1 cells and the induction of IFN-γ by various immune cells including NK cells. During the progress of metabolic syndrome, abnormal metabolites, such as oxidized LDL and hyperglycemia activate the TLR4 and/or TLR2-mediated pathways [51][52], potentially leading to the production of IL-12 and/or IL-15. Under these conditions, IL-18 might activate NK cells and/or Th1 cells to produce large amounts of IFN-γ and/or TNF-α [1][2][14][53][54].
2.5. IL-18 in Cancer
IL-18 activates NK cells to produce IFN-γ and enhance cytotoxicity against tumor cells in synergy with IL-12 [1][53][54][55]. Because NK cells, and recently identified innate lymphoid cells, are well-established tumor-killing cells [56], many researchers have addressed whether IL-18 therapy rescues cancer expansion [57][58][59]. Here, we describe two recent theories on the beneficial roles of IL-18 in protecting against cancer. One is the establishment of cancer therapy by IL-18-activated human γδT cells. The other topic is the importance of fungi in microbiota for protection against colitis-associated colorectal cancer by inducing IL-18.
2.5.1. IL-18 Robustly Expands Human γδT Cells
γδT cells have several innate cell-like properties [60][61]. For example, similar to αβT cells, γδT cells are activated upon T cell receptor (TCR) engagement. Whereas the TCR-mediated activation of αβT cells occurs in an MHC-restricted manner, the TCR engagement of γδT cells is independent of the MHC. To exert their biological function, naïve αβT cells require the appropriate differentiation into effector T cells, whereas γδT cells, including NK cells, can rapidly produce large amounts of cytokines and kill tumor cells. Indeed, γδT cells are well documented to exert tumoricidal activity [60][61][62]. However, low numbers of γδT ells are present in the peripheral blood of humans. Therefore, the bottleneck for the development of γδT cell-mediated cancer therapy has been the lack of an established method suitable for the efficient and safe expansion of γδT cells. Recently, Okamura’s group reported a protocol to obtain high numbers of γδT cells using IL-18 [63][64][65][66]. The incubation of human PBMCs including 1%–2% γδT cells with γδT cell Ag and IL-2 and IL-18, induced the proliferation of γδT cells, but not αβT cells, by approximately several thousand-fold in a 2-week culture [63][64][65][66] (Figure 6A). They used zoledronate as an activator of endogenous γδT cell Ag, which induces the accumulation of intermediate isopentenyl pyrophosphate, an endogenous γδT cell Ag, by blocking farnesyl pyrophosphate synthase in human monocytes [62][67][68]. The depletion of monocytes from PBMCs prevented the expansion of γδT cells [64]. Intriguingly, the IL-18-mediated expansion of human γδT cells requires CD56+CD11c+cells [63][64], initially termed NK-like dendritic cells (NKDCs) [69][70]. Indeed, CD56intCD11c+ cells in PBMCs co-cultured with monocytes in the presence of IL-12 and IL-18 robustly expanded and differentiated into CD56brightCD11c+ cells [63]. However, how CD56brightCD11c+ cells contribute to the expansion of γδT cells remains unknown. They also demonstrated that combination therapy with IL-18 and immune-checkpoint therapy with anti-PD-L1 and/or anti-CTLA4 mAb, synergistically prevented the mortality of mice harboring various tumor cell lines [71]. Combination therapy with IL-18 induced the expansion of precursor mature NK cells (counter cells of human CD56+CD11c+ cells) but did not affect regulatory T cells. The in vivo depletion of precursor mature NK cells or CD8+ T cells abrogated these therapeutic effects [71]. Therefore, IL-18 in combination with immune-checkpoint therapy might be a potential treatment for the early stages of cancer in humans.
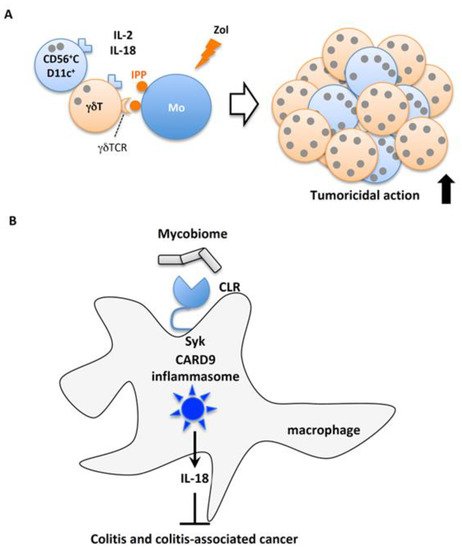
Figure 6. IL-18 protection against cancer. (A) Robust proliferation of tumoricidal human γδT cells. IL-18 in combination with IL-2 activates and induces the proliferation of human CD56+CD11c+ precursor NK cells, which in turn robustly proliferate and activate γδT cells stimulated with γδT cell antigen produced by zoledronate (Zol)-treated monocytes (Mo). (B) Involvement of CLRs-mediated CARD9 inflammasome activation in the induction of colon cancer. Fungi in the intestinal microbial flora activate macrophages through C-type lectin receptors (CLRs), which promote Syk to activate caspase-1 via the CARD9 inflammasome. The resultant IL-18 is required for protection against colitis and colitis-associated cancer.
2.5.2. Mycobiome-Mediated IL-18 Protects Against Colitis-Associated Colorectal Cancer
A recent report confirmed the anti-cancer effect of IL-18 released from macrophages in response to commensal fungi on colitis-associated cancer [72]. The microbiome contains fungi as well as bacteria and other microorganisms [73]. C-type lectin receptors including Dectin-1, Dectin-2, Dectin-3, and Mincle are expressed on host cells and recognize β-glucan and α-mannans expressed by fungi [74][75][76][77]. Upon ligation with their ligands, C-type lectin receptors recruit Syk kinase, followed by NF-κB and MAPK signaling by assembling the CARD9/MALT/BCL10 complex [78]. Malik et al. reported that the recognition of commensal fungi by C-type lectin receptor induced Syk-dependent CARD9 inflammasome activation induced the release of mature IL-18 [72] (Figure 6B). Card9−/− mice and mice selectively deficient for Syk in myeloid cells were predisposed to azoxymethane (AOM)/DSS-induced colitis-associated colorectal cancer, concomitant with reduced mature IL-18 in colon explants and an impaired accumulation of anti-tumorigenic T cells in the colon. Exogenous IL-18 prevented these mutant mice from colorectal cancer and restored the migration of anti-tumorigenic T cells. Of note, the administration of antifungal drugs rendered wild type mice highly susceptible to AOM/DSS-induced colitis-associated colorectal cancer, and supplementation with IL-18 rescued their predisposition to colorectal cancer [72]. These observations suggest the careful per os treatment of IBD patients with antifungal drugs might be of benefit. The depletion of mycobiota by antifungal drugs might initiate and/or promote colorectal cancer in IBD patients.
3. IL-18 as A Therapeutic Target
Because of the strong proinflammatory activity of IL-18, many researchers are investigating IL-18 as a therapeutic target for the treatment of inflammatory diseases. To neutralize IL-18, IL-18 BP or anti-IL-18 Ab formulations were devised and clinical trials have been conducted to verify its safety and efficacy [79][80]. Clinical trials are underway to investigate the treatment of adult-onset Still’s disease and NLRC4-related macrophage activation syndrome (inflammatory diseases associated with high plasma IL-18 levels) using IL-18BP [81][82][83] (ClinicalTrials.gov Identifier: NCT 02398435, NCT 03113760).
In addition, the immunostimulatory effects of IL-18 have been investigated for treatments. The first attempts to administer IL-18 to cancer patients showed that its toxicity was generally mild-to-moderate [84]. For optimal cancer therapy, combination with other therapies is being considered. Anti-CD20 Ab is used to treat CD20 positive B cell lymphoma. Clinical studies using IL-18 with an anti-CD20 Ab are underway, and it was reported that the effect of anti-CD20 Ab was enhanced by the administration of IL-18 [85].
Although it is still at the stage of animal experiments, a new treatment method using IL-18 for cancer treatment has been studied. Recently, immune-checkpoint therapy by the neutralization of PD-1 or CTLA4 has dramatically improved cancer treatment. Combination therapy with IL-18 and an immune-checkpoint inhibitor synergistically reduced mortality in mice harboring various tumor cell lines. Therefore, IL-18 in combination with immune-checkpoint therapy might be a potential treatment for the early stages of cancer in humans [71]. Furthermore, chimeric antigen receptor (CAR) T cells artificially expressing a cancer antigen-specific TCR were effective treatments for B cell lymphoma and leukemia [86][87]. Studies on the effect on tumors of expressing IL-18 in CAR T cells in mice, demonstrated that IL-18 enhanced the antitumor effect [88][89]. These new therapy methods are expected to be applied to humans in the future and to save those suffering from cancer.
This entry is adapted from the peer-reviewed paper 10.3390/ijms20030649
References
- Okamura, H.; Tsutsui, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature 1995, 378, 88–91.
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 2001, 19, 423–474.
- Ushio, S.; Namba, M.; Okura, T.; Hattori, K.; Nukada, Y.; Akita, K.; Tanabe, F.; Konishi, K.; Micallef, M.; Fujii, M.; et al. Cloning of the cDNA for human IFN-g-inducing factor, expression in Escherichia coli, and studies on the biologic activities of the protein. J. Immunol. 1996, 156, 4274–4279.
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of interferon-g inducing factor mediated by interleukin-1b converting enzyme. Science 1997, 275, 206–209.
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265.
- Adachi, O.; Kawai, T.; Takeda, K.; Matsumoto, M.; Tsutsui, H.; Sakagami, M.; Nakanishi, K.; Akira, S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998, 9, 143–150.
- Nakanishi, K. Unique Action of Interleukin-18 on T Cells and Other Immune Cells. Front. Immunol. 2018, 9, 763.
- Yoshimoto, T.; Nakanishi, K.; Hirose, K.; Hiroishi, K.; Okamura, H.; Takemoto, Y.; Kanamaru, A.; Hada, T.; Tamura, T.; Kakishita, E.; et al. High serum IL-6 level reflects susceptible status of the host to endotoxin and IL-1/tumor necrosis factor. J. Immunol. 1992, 148, 3596–3603.
- Tsutsui, H.; Matsui, K.; Okamura, H.; Nakanishi, K. Pathophysiological roles of interleukin-18 for inflammatory liver diseases. Immunol. Rev. 2000, 174, 192–209.
- Kawa, K.; Tsutsui, H.; Uchiyama, R.; Kato, J.; Matsui, K.; Iwakura, Y.; Matsumoto, T.; Nakanishi, K. IFN-g is a master regulator of endotoxin shock syndrome in mice primed with heat-killed Propionibacterium acnes. Int. Immunol. 2010, 22, 157–166.
- Tsutsui, H.; Kayagaki, N.; Kuida, K.; Nakano, H.; Hayashi, N.; Takeda, K.; Matsui, K.; Kashiwamura, S.-I.; Hada, T.; Akira, S.; et al. Caspase-1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity 1999, 11, 359–367.
- Imamura, M.; Tsutsui, H.; Yasuda, K.; Uchiyama, R.; Yumikura-Futatsugi, S.; Mitani, K.; Hayashi, S.; Akira, S.; Taniguchi, S.; Van Rooijen, N.; et al. Contribution of TIR domain-containing adapter inducing IFN-β-mediated IL-18 release to LPS-induced liver injury in mice. J. Hepatol. 2009, 51, 333–341.
- Seki, E.; Tsutsui, H.; Nakano, H.; Tsuji, N.M.; Hoshino, K.; Adachi, O.; Adachi, K.; Futatsugi, S.; Kuida, K.; Takeuchi, O.; et al. LPS-induced IL-18 secretion from murine Kupffer cells independently of MyD88 that is critically involved in induction of production of IL-12 and IL-1β. J. Immunol. 2001, 166, 2651–2657.
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: Synergism with IL-18 for IFN-g production. J. Immunol. 1998, 161, 3400–3407.
- Yoshimoto, T.; Mizutani, H.; Tsutsui, H.; Noben-Trauth, N.; Yamanaka, K.; Tanaka, M.; Izumi, S.; Okamura, H.; Paul, W.E.; Nakanishi, K. IL-18 induction of IgE: Dependence on CD4+ T cells, IL-4 and STAT6. Nat. Immunol. 2000, 1, 132–137.
- Konishi, H.; Tsutsui, H.; Murakami, T.; Yumikura-Futatsugi, S.; Yamanaka, K.; Tanaka, M.; Iwakura, Y.; Suzuki, N.; Takeda, K.; Akira, S.; et al. IL-18 contributes to the spontaneous development of atopic dermatitis-like inflammatory skin lesion independently of IgE/stat6 under specific pathogen-free conditions. Proc. Natl. Acad. Sci. USA 2002, 99, 11340–11345.
- Tsutsui, H.; Yoshimoto, T.; Hayashi, N.; Mizutani, H.; Nakanishi, K. Induction of allergic inflammation by interleukin-18 in experimental animal models. Immunol. Rev. 2004, 202, 115–138.
- Sugimoto, T.; Ishikawa, Y.; Yoshimoto, T.; Hayashi, B.; Fujimoto, J.; Nakanishi, K. IL-18 acts on memory Th1 cells to induce airway inflammatin and hyperresponsiveness in a naive host mouse. J. Exp. Med. 2004, 199, 535–545.
- Hayashi, N.; Yoshimoto, T.; Izuhara, K.; Matsui, K.; Tanaka, T.; Nakanishi, K. T helper 1 cells stimulated with ovalbunmin and IL-18 induce airway hyperresponsiveness and lung fibrosis by IFN-g and IL-13 production. Proc. Natl. Acad. Sci. USA 2007, 104, 14765–14770.
- Ishikawa, Y.; Yoshimoto, T.; Nakanishi, K. Contribution of IL-18-induced innate T cell activation to airway inflammation with mucus hypersecretion and airway hyperresponsiveness. Int. Immunol. 2006, 18, 847–855.
- Kondo, Y.; Yoshimoto, T.; Yasuda, K.; Futatsugi-Yumikura, S.; Morimoto, M.; Hayashi, N.; Hoshino, T.; Fujimoto, J.; Nakanishi, K. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lung in the absence of adaptive immune system. Int. Immunol. 2008, 20, 791–800.
- Wu, H.; Craft, M.L.; Wang, P.; Wyburn, K.R.; Chen, G.; Ma, J.; Hambly, B.; Chadban, S.J. IL-18 contributes to renal damage after ischemia-reperfusion. J. Am. Soc. Nephrol. 2008, 19, 2331–2341.
- Faubel, S.; Ljubanovic, D.; Reznikov, L.; Somerset, H.; Dinarello, C.A.; Edelstein, C.L. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int. 2004, 66, 2202–2213.
- Bani-Hani, A.H.; Leslie, J.A.; Asanuma, H.; Dinarello, C.A.; Campbell, M.T.; Meldrum, D.R.; Zhang, H.; Hile, K.; Meldrum, K.K. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int. 2009, 76, 500–511.
- Masood, H.; Che, R.; Zhang, A. Inflammasomes in the Pathophysiology of Kidney Diseases. Kidney Dis. (Basel) 2015, 1, 187–193.
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology (Carlton) 2016, 21, 736–744.
- Purves, J.T.; Hughes, F.M., Jr. Inflammasomes in the urinary tract: A disease-based review. Am. J. Physiol. Renal Physiol. 2016, 311, F653–F662.
- Hamilton, C.; Tan, L.; Miethke, T.; Anand, P.K. Immunity to uropathogens: The emerging roles of inflammasomes. Nat. Rev. Urol. 2017, 14, 284–295.
- D’Amico, G. Natural history of idiopathic IgA nephropathy: Role of clinical and histological prognostic factors. Am. J. Kidney Dis. 2000, 36, 227–237.
- Koyama, A.; Igarashi, M.; Kobayashi, M. Natural history and risk factors for immunoglobulin A nephropathy in Japan. Research Group on Progressive Renal Diseases. Am. J. Kidney Dis. 1997, 29, 526–532.
- Shi, B.; Ni, Z.; Cao, L.; Zhou, M.; Mou, S.; Wang, Q.; Zhang, M.; Fang, W.; Yan, Y.; Qian, J. Serum IL-18 is closely associated with renal tubulointerstitial injury and predicts renal prognosis in IgA nephropathy. Mediat. Inflamm. 2012, 2012, 728417.
- Liu, D.; Xu, M.; Ding, L.H.; Lv, L.L.; Liu, H.; Ma, K.L.; Zhang, A.H.; Crowley, S.D.; Liu, B.C. Activation of the Nlrp3 inflammasome by mitochondrial reactive oxygen species: A novel mechanism of albumin-induced tubulointerstitial inflammation. Int J. Biochem. Cell Biol. 2014, 57, 7–19.
- Melnikov, V.Y.; Ecder, T.; Fantuzzi, G.; Siegmund, B.; Lucia, M.S.; Dinarello, C.A.; Schrier, R.W.; Edelstein, C.L. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J. Clin. Investig. 2001, 107, 1145–1152.
- Melnikov, V.Y.; Faubel, S.; Siegmund, B.; Lucia, M.S.; Ljubanovic, D.; Edelstein, C.L. Neutrophil-independent mechanisms of caspase-1- and IL-18-mediated ischemic acute tubular necrosis in mice. J. Clin. Investig. 2002, 110, 1083–1091.
- Parikh, C.R.; Jani, A.; Melnikov, V.Y.; Faubel, S.; Edelstein, C.L. Urinary interleukin-18 is a marker of human acute tubular necrosis. Am. J. Kidney Dis. 2004, 43, 405–414.
- Parikh, C.R.; Mishra, J.; Thiessen-Philbrook, H.; Dursun, B.; Ma, Q.; Kelly, C.; Dent, C.; Devarajan, P.; Edelstein, C.L. Urinary IL-18 is an early predictive biomarker of acute kidney injury after cardiac surgery. Kidney Int. 2006, 70, 199–203.
- Schrezenmeier, E.V.; Barasch, J.; Budde, K.; Westhoff, T.; Schmidt-Ott, K.M. Biomarkers in acute kidney injury—Pathophysiological basis and clinical performance. Acta Physiol. (Oxf.) 2017, 219, 554–572.
- Coca, S.G.; Yusuf, B.; Shlipak, M.G.; Garg, A.X.; Parikh, C.R. Long-term risk of mortality and other adverse outcomes after acute kidney injury: A systematic review and meta-analysis. Am. J. Kidney Dis. 2009, 53, 961–973.
- Li, S.; Krawczeski, C.D.; Zappitelli, M.; Devarajan, P.; Thiessen-Philbrook, H.; Coca, S.G.; Kim, R.W.; Parikh, C.R.; Consortium, T.-A. Incidence, risk factors, and outcomes of acute kidney injury after pediatric cardiac surgery: A prospective multicenter study. Crit. Care Med. 2011, 39, 1493–1499.
- Zappitelli, M.; Greenberg, J.H.; Coca, S.G.; Krawczeski, C.D.; Li, S.; Thiessen-Philbrook, H.R.; Bennett, M.R.; Devarajan, P.; Parikh, C.R.; Translational Research Investigating Biomarker Endpoints in Acute Kidney Injury (TRIBE-AKI) Consortium. Association of definition of acute kidney injury by cystatin C rise with biomarkers and clinical outcomes in children undergoing cardiac surgery. JAMA Pediatr. 2015, 169, 583–591.
- Coca, S.G.; Garg, A.X.; Thiessen-Philbrook, H.; Koyner, J.L.; Patel, U.D.; Krumholz, H.M.; Shlipak, M.G.; Parikh, C.R.; Consortium, T.-A. Urinary biomarkers of AKI and mortality 3 years after cardiac surgery. J. Am. Soc. Nephrol. 2014, 25, 1063–1071.
- Feltes, C.M.; Van Eyk, J.; Rabb, H. Distant-organ changes after acute kidney injury. Nephron Physiol. 2008, 109, p80–84.
- Kelly, K.J. Distant effects of experimental renal ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2003, 14, 1549–1558.
- Parikh, C.R.; Puthumana, J.; Shlipak, M.G.; Koyner, J.L.; Thiessen-Philbrook, H.; McArthur, E.; Kerr, K.; Kavsak, P.; Whitlock, R.P.; Garg, A.X.; et al. Relationship of Kidney Injury Biomarkers with Long-Term Cardiovascular Outcomes after Cardiac Surgery. J. Am. Soc. Nephrol. 2017, 28, 3699–3707.
- Hulthe, J.; McPheat, W.; Samnegård, A.; Tornvall, P.; Hamsten, A.; Eriksson, P. Plasma interleukin (IL)-18 concentrations is elevated in patients with previous myocardial infarction and related to severity of coronary atherosclerosis independently of C-reactive protein and IL-6. Atherosclerosis 2006, 188, 4450–4454.
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Leséche, G.; Chvatchko, Y.; Tedgui, A. Expression of Interleukin-18 in human atherosclerotic plaque and relation to plaque instability. Circulation 2001, 104, 1598–1603.
- Blankenberg, S.; Tiret, L.; Bickel, C.; Peetz, D.; Cambien, F.; Meyer, J.; Rupprecht, H.J.; AtheroGene, I. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation 2002, 106, 24–30.
- Blankenberg, S.; Luc, G.; Ducimetiére, P.; Arveiler, D.; Ferriéres, J.; Amouyel, P.; Evans, A.; Cambien, F.; Tiret, L. Interleukin-18 and the risk of coronary heart disease in european men: The prospective epidemiological study of myocardial infarction (PRIME). Circulation 2003, 108, 2453–2459.
- Jefferis, B.J.M.H.; Papacosta, O.; Owen, C.G.; Toya Wannamethee, S.; Humphries, S.E.; Woodward, M.; Lennon, L.T.; Thomson, A.; Welsh, P.; Rumley, A.; et al. Interleukin 18 and coronary heart disease: Prospective study and systematic review. Atherosclerosis 2011, 217, 227–233.
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147.
- Stewart, C.R.; stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.C.; Halle, A.; Ryaner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161.
- Dasu, M.R.; Devaray, S.; Zhao, L.; Hwang, D.H.; Jialal, I. High Glucose Induces Toll-Like Receptor Expression in Human Monocytes. Diabetes 2008, 57, 3090–3098.
- Hyodo, Y.; Matsui, K.; Hayashi, N.; Tsutsui, H.; Kashiwamura, S.; Yamauchi, H.; Hiroishi, K.; Takeda, K.; Tagawa, Y.; Iwakura, Y.; et al. IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J. Immunol. 1999, 162, 1662–1668.
- Takeda, K.; Tsutsui, H.; Yoshimoto, T.; Adachi, O.; Yoshida, N.; Kishimoto, T.; Okamura, H.; Nakanishi, K.; Akira, S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 1998, 8, 383–390.
- Okamura, H.; Kashiwamura, S.; Tsutsui, H.; Yoshimoto, T.; Nakanishi, K. Regulation of interferon-g production by IL-12 and IL-18. Curr. Opin. Immunol. 1998, 10, 259–264.
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688.
- Granzin, M.; Wagner, J.; Kohl, U.; Cerwenka, A.; Huppert, V.; Ullrich, E. Shaping of Natural Killer Cell Antitumor Activity by Ex Vivo Cultivation. Front. Immunol. 2017, 8, 458.
- Park, S.; Cheon, S.; Cho, D. The Dual Effects of Interleukin-18 in Tumor Progression. Cell. Mol. Immunol 2007, 4, 329–335.
- Fabbi, M.; Carbotti, G.; Ferrini, S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. J. Leukoc. Biol. 2015, 97, 665–675.
- Bonneville, M.; O’Brien, R.L.; Born, W.K. γδ T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478.
- Fisher, J.; Anderson, J. Engineering Approaches in Human Gamma Delta T Cells for Cancer Immunotherapy. Front. Immunol. 2018, 9, 1409.
- Locatelli, F.; Merli, P.; Rutella, S. At the Bedside: Innate immunity as an immunotherapy tool for hematological malignancies. J. Leukoc. Biol. 2013, 94, 1141–1157.
- Li, W.; Kubo, S.; Okuda, A.; Yamamoto, H.; Ueda, H.; Tanaka, T.; Nakamura, H.; Yamanishi, H.; Terada, N.; Okamura, H. Effect of IL-18 on Expansion of gd T Cells Stimulated by Zoledronate and IL-2. J. Immunother. 2010, 33, 287–296.
- Tsuda, J.; Li, W.; Yamanishi, H.; Yamamoto, H.; Okuda, A.; Kubo, S.; Ma, Z.; Terada, N.; Tanaka, Y.; Okamura, H. Involvement of CD56brightCD11c+ Cells in IL-18–Mediated Expansion of Human gd T Cells. J. Immunol. 2011, 186, 2003–2012.
- Li, W.; Okuda, A.; Yamamoto, H.; Yamanishi, K.; Terada, N.; Yamanishi, H.; Tanaka, Y.; Okamura, H. Regulation of Development of CD56brightCD11c+ NK-like Cells with Helper Function by IL-18. PLoS ONE 2013, 8, e82586.
- Sugie, T.; Murata-Hirai, K.; Iwasaki, M.; Morita, C.T.; Li, W.; Okamura, H.; Minato, N.; Toi, M.; Tanaka, Y. Zoledronic acid-induced expansion of gammadelta T cells from early-stage breast cancer patients: Effect of IL-18 on helper NK cells. Cancer Immunol. Immunother. 2013, 62, 677–687.
- Gober, H.-J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T Cell Receptor gamma delta Cells Recognize Endogenous Mevalonate Metabolites in Tumor Cells. J. Exp. Med. 2003, 197, 163–168.
- Roelofs, A.J.; Jauhiainen, M.; Monkkonen, H.; Rogers, M.J.; Monkkonen, J.; Thompson, K. Peripheral blood monocytes are responsible for gammadelta T cell activation induced by zoledronic acid through accumulation of IPP/DMAPP. Br. J. Haematol. 2009, 144, 245–250.
- Guimont-Desrochers, F.; Boucher, G.; Dong, Z.; Dupuis, M.; Veillette, A.; Lesage, S. Redefining interferon-producing killer dendritic cells as a novel intermediate in NK-cell differentiation. Blood 2012, 119, 4349–4357.
- Guimont-Desrochers, F.; Lesage, S. Revisiting the prominent anti-tumoral potential of pre-mNK cells. Front. Immunol. 2013, 4, Article–446.
- Ma, Z.; Li, W.; Yoshiya, S.; Xu, Y.; Hata, M.; El-Darawish, Y.; Markova, T.; Yamanishi, K.; Yamanishi, H.; Tahara, H.; et al. Augmentation of Immune Checkpoint Cancer Immunotherapy with IL18. Clin. Cancer Res. 2016, 22, 2969–2980.
- Malik, A.; Sharma, D.; Malireddi, R.K.S.; Guy, C.S.; Chang, T.-C.; Olsen, S.R.; Neale, G.A.; Vogel, P.; Kanneganti, T.-D. SYK-CARD9 Signaling Axis Promotes Gut Fungi-Mediated Inflammasome Activation to Restrict Colitis and Colon Cancer. Immunity 2018, 49, 515–530.
- Sartor, R.B.; Wu, G.D. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology 2017, 152, 327–339.
- Yamasaki, S.; Matsumoto, M.; Takeuchi, O.; Matsuzawa, T.; Ishikawa, E.; Sakuma, M.; Tateno, M.; Uno, J.; Hirabayashi, J.; Mikami, Y.; et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc. Natl. Acad. Sci. USA 2009, 106, 1897–1902.
- Saijo, S.; Fujikado, N.; Furuta, T.; Chung, S.-h.; Kotaki, H.; seki, K.; Sudo, K.; Akira, S.; Adachi, Y.; Ohno, N.; et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol. 2007, 8, 39–46.
- Saijo, S.; Ikeda, S.; Yamabe, K.; Kakuta, S.; Ishigame, H.; Akitsu, A.; Fujikado, N.; Kusaka, T.; Kubo, S.; Chung, S.-h.; et al. Dectin-2 Recognition of a-Mannans and Induction of Th17 Cell Differentiation Is Essential for Host Defense against Candida albicans. Immunity 2010, 32, 681–691.
- Zhu, L.-L.; Zhao, X.-Q.; Jiang, C.; You, Y.; Chen, X.-P.; Jiang, Y.-Y.; Jia, X.-M.; Lin, X. C-Type Lectin Receptors Dectin-3 and Dectin-2 Form a Heterodimeric Pattern-Recognition Receptor for Host Defense against Fungal Infection. Immunity 2013, 39, 324–334.
- Drummond, R.A.; Franco, L.M.; Lionakis, M.S. Human CARD9: A Critical Molecule of Fungal Immune Surveillance. Front. Immunol. 2018, 9, 1836.
- Tak, P.P.; Bacchi, M.; Bertolino, M. Pharmacokinetics of IL-18 binding protein in healthy volunteers and subjects with rheumatoid arthritis or plaque psoriasis. Eur. J. Drug Metab. Pharmacokinet. 2006, 31, 109–116.
- Robertson, M.J.; Mier, J.W.; Logan, T.; Atkins, M.; Koon, H.; Koch, K.M.; Kathman, S.; Pandite, L.N.; Oei, C.; Kirby, L.C.; et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin. Cancer Res. 2006, 12, 4265–4273.
- Colafrancesco, S.; Priori, R.; Alessandri, C.; Perricone, C.; Pendolino, M.; Picarelli, G.; Valesini, G. IL-18 Serum Level in Adult Onset Still’s Disease: A Marker of Disease Activity. Int. J. Inflamm. 2012, 2012, 156890.
- Gabay, C.; Fautrel, B.; Rech, J.; Spertini, F.; Feist, E.; Kotter, I.; Hachulla, E.; Morel, J.; Schaeverbeke, T.; Hamidou, M.A.; et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann. Rheum. Dis. 2018, 77, 840–847.
- Canna, S.W.; Girard, C.; Malle, L.; de Jesus, A.; Romberg, N.; Kelsen, J.; Surrey, L.F.; Russo, P.; Sleight, A.; Schiffrin, E.; et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J. Allergy Clin. Immunol. 2017, 139, 1698–1701.
- Robertson, M.J.; Kirkwood, J.M.; Logan, T.F.; Koch, K.M.; Kathman, S.; Kirby, L.C.; Bell, W.N.; Thurmond, L.M.; Weisenbach, J.; Dar, M.M. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin Cancer Res. 2008, 14, 3462–3469.
- Robertson, M.J.; Kline, J.; Struemper, H.; Koch, K.M.; Bauman, J.W.; Gardner, O.S.; Murray, S.C.; Germaschewski, F.; Weisenbach, J.; Jonak, Z.; et al. A dose-escalation study of recombinant human interleukin-18 in combination with rituximab in patients with non-Hodgkin lymphoma. J. Immunother. 2013, 36, 331–341.
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733.
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517.
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033.
- Kunert, A.; Chmielewski, M.; Wijers, R.; Berrevoets, C.; Abken, H.; Debets, R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 2017, 7, e1378842.
This entry is offline, you can click here to edit this entry!