The heart is a metabolic omnivore that combusts a considerable amount of energy substrates, mainly long-chain fatty acids (FAs) and others such as glucose, lactate, ketone bodies, and amino acids. There is emerging evidence that muscle-type continuous capillaries comprise the rate-limiting barrier that regulates FA uptake into cardiomyocytes. The transport of FAs across the capillary endothelium is composed of three major steps—the lipolysis of triglyceride on the luminal side of the endothelium, FA uptake by the plasma membrane, and intracellular FA transport by cytosolic proteins. In the heart, impaired trans-endothelial FA (TEFA) transport causes reduced FA uptake, with a compensatory increase in glucose use. In most cases, mice with reduced FA uptake exhibit preserved cardiac function under unstressed conditions. When the workload is increased, however, the total energy supply relative to its demand (estimated with pool size in the tricarboxylic acid (TCA) cycle) is significantly diminished, resulting in contractile dysfunction. The supplementation of alternative fuels, such as medium-chain FAs and ketone bodies, at least partially restores contractile dysfunction, indicating that energy insufficiency due to reduced FA supply is the predominant cause of cardiac dysfunction.
- cardiac metabolism
- fatty acid
- capillary endothelium
- trans-endothelial fatty acid transport
- contractile function
1. Mechanisms of FA Uptake by the Heart
1.1. Source of Long-Chain Fatty Acids
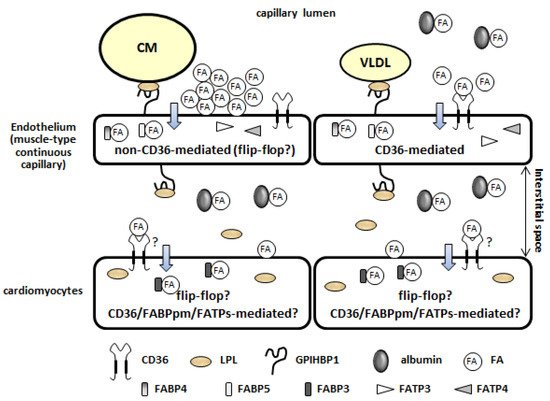
1.2. Lipolysis of TG Contained in TG-Rich Lipoproteins on the Luminal Side of the Capillary Endothelium
1.3. Fatty Acid Uptake by the Plasma Membrane of the Capillary Endothelium (Non-CD36-Mediated and CD36-Mediated Pathways)
1.4. Intracellular Fatty Acid Transport through the Capillary Endothelium
1.5. Fatty Acid Uptake by Cardiomyocytes
2. Molecular Mechanisms Underlying the Induction of Genes Associated with Trans-Endothelial Fatty Acid Transport
Recent studies have revealed that the expression of genes associated with TEFA transport is regulated by several ligands, receptors, and transcription factors (Table 1) [21][22][23][24]. It is likely that these systems can be roughly divided into two groups according to their target genes. One includes the peroxisome proliferator-activated receptor γ (PPARγ), mesodermal homeobox-2/transcription factor 15 (Meox2/Tcf15), Notch signaling, and the apelin/apelin receptor (APLNR), and it mainly controls the expression of CD36, FABPs, and GPIHBP1. The other is a group that includes the VEGF-B/VEGF receptor (VEGFR), angiopoietin-like 2 (ANGPTL2), and 3-hydroxyisobutyrate (3-HIB), and it regulates the expression/function of FATP3/4 (Table 1). Although impairments of the systems influence both local and systemic metabolism, cardiac metabolism seems to only be affected by PPARγ, Meox2/Tcf15, Notch signaling, and VEGF-B/VEGFR (Table 1) [21][22][23][24]. The trans-endothelial transport of other substrates and molecules (e.g., lipoproteins, lipoprotein lipase, glucose, and insulin) and endothelium-derived metabolic regulators (e.g., nitric oxide, extracellular matrix proteins, hormones, growth factors, and enzymes) is described elsewhere [21][22][24].
| Ligand | Receptor/Transcription Factor | Target Genes | Target Tissues Influenced by the System | Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PPARγ | CD36 | FABP4 | FABP5 | LPL | GPIHBP1 | ANGPTL4 | LIPG | FATP3 | FATP4 | ||||
| PPARγ | ⚪ | ⚪ | ⚪ | heart, skeletal muscle, adipose tissue | [25][26][27] | ||||||||
| Meox2/Tcf15 | ⚪ | ⚪ | ⚪ | ⚪ | ⚪ | ⚪ | heart | [28] | |||||
| Dll4 | Notch1/N1-ICD/Rbp-jκ | independent | ⚪ | ⚪ | ⚪ | ⚫ | ⚪ | heart, skeletal muscle | [29][30] | ||||
| Apelin | APLNR/phosphorylation of FOXO1 | ⚫ | skeletal muscle | [31] | |||||||||
| VEGF-B | VEGFR/NPR1 | ⚪ | ⚪ | heart, BAT, skeletal muscle | [16] | ||||||||
| ANGPTL2 | integrin α5β1 | ⚪ | ⚪ | subcutaneous adipose tissue | [32] | ||||||||
| 3-HIB | ⚪* | ⚪* | skeletal muscle | [33] | |||||||||
2.1. Peroxisome Proliferator-Activated Receptor γ
2.2. Mesodermal Homeobox-2/Transcription Factor 15
2.3. Notch Signaling
| Target Genes | Deficient Site | Inducible Knockout | VLDL-TG Uptake | FA Uptake | Glucose Uptake | Glut1/4 | Ketonein Serum | Contractile Performance In Vivo Estimated by Echocardiography | Reference |
|---|---|---|---|---|---|---|---|---|---|
| LPL (functions at luminal side of capillary) | cardiomyocyte | ↓ | ↑ | ↑ | ↑ | ↓ aged | [37] | ||
| cardiomyocyte | ⚪ | ↓ | [38] | ||||||
| CD36 | whole | ↓ | ↑ | ↑ | ↑ | intact | [39][40][41][42] | ||
| whole | ↓ | ↑ | prevention from age-induced cardiomyopathy | [42] | |||||
| endothelium | ↓ | ↑ | ↑ | not available | [11] | ||||
| FABP4/5 | whole | ↓ | ↑ | ↑ | ↑ | intact | [13][43] | ||
| Meox2+/−:Tcf15+/− | endothelium: whole | ↓ | ↑ | ↓ aged | [28] | ||||
| Rbp-jκ (Notch signal) | endothelium | ⚪ | ↓ | ↑ | ↓ | ↓↓ | [29] | ||
| PPARγ | endothelium | →↓ | → | intact (personal observation) | [25] | ||||
| VEGF-B | whole | ↓ | ↑ | ↑ | not available | [16] | |||
| FABP3 | whole | ↓ | ↑ | → | ↑ | not available | [44][45] | ||
| CD36 | cardiomyocyte | → | → | not available | [11] | ||||
| cardiomyocyte | ⚪ | ↓ (ex vivo) | ↑ (ex vivo) | intact | [46][47] |
2.4. Apelin/Apelin Receptor/Forkhead Box O1
2.5. Angiopoietin-Like 2/Integrin α5β1
2.6. 3-Hydroxyisobutyrate
3. Association between In Vivo Cardiac Metabolism and Contractile Function in Mice with Reduced Fatty Acid Uptake
3.1. Limitation of Experiments with Ex Vivo Perfused Hearts
3.2. In Vivo Cardiac Metabolism and Contractile Function in Mice with Reduced Trans-Endothelial Fatty Acid Transport under Unstressed Conditions
3.3. In Vivo Cardiac Metabolism in CD36 KO Mice under Unstressed Conditions
3.4. In Vivo Contractile Dysfunction in Mice with Reduced Trans-Endothelial Fatty Acid Transport under an Increased Afterload
3.5. Pool Size in the TCA Cycle as a Useful Marker for Energy Status
3.6. Mechanism Underlying the Enhancement of Glycolytic Flux in the Hearts of Mice with Reduced Fatty Acid Uptake
This entry is adapted from the peer-reviewed paper 10.3390/metabo11120889
References
- Basu, D.; Goldberg, I.J. Regulation of lipoprotein lipase-mediated lipolysis of triglycerides. Curr. Opin. Lipidol. 2020, 31, 154–160.
- Young, S.G.; Fong, L.G.; Beigneux, A.P.; Allan, C.M.; He, C.; Jiang, H.; Nakajima, K.; Meiyappan, M.; Birrane, G.; Ploug, M. GPIHBP1 and Lipoprotein Lipase, Partners in Plasma Triglyceride Metabolism. Cell Metab. 2019, 30, 51–65.
- Evans, R.D.; Hauton, D. The role of triacylglycerol in cardiac energy provision. Biochim. Biophys. Acta 2016, 1860, 1481–1491.
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258.
- Van der Vusse, G.J.; van Bilsen, M.; Glatz, J.F. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc. Res. 2000, 45, 279–293.
- Adeyo, O.; Goulbourne, C.N.; Bensadoun, A.; Beigneux, A.P.; Fong, L.G.; Young, S.G. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 and the intravascular processing of triglyceride-rich lipoproteins. J. Intern. Med. 2012, 272, 528–540.
- Takahashi, S.; Sakai, J.; Fujino, T.; Hattori, H.; Zenimaru, Y.; Suzuki, J.; Miyamori, I.; Yamamoto, T.T. The very low-density lipoprotein (VLDL) receptor: Characterization and functions as a peripheral lipoprotein receptor. J. Atheroscler. Thromb. 2004, 11, 200–208.
- Wyne, K.L.; Pathak, K.; Seabra, M.C.; Hobbs, H.H. Expression of the VLDL receptor in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 407–415.
- Abumrad, N.A.; Goldberg, I.J. CD36 actions in the heart: Lipids, calcium, inflammation, repair and more? Biochim. Biophys. Acta 2016, 1860, 1442–1449.
- Bharadwaj, K.G.; Hiyama, Y.; Hu, Y.; Huggins, L.A.; Ramakrishnan, R.; Abumrad, N.A.; Shulman, G.I.; Blaner, W.S.; Goldberg, I.J. Chylomicron- and VLDL-derived lipids enter the heart through different pathways: In vivo evidence for receptor- and non-receptor-mediated fatty acid uptake. J. Biol. Chem. 2010, 285, 37976–37986.
- Son, N.H.; Basu, D.; Samovski, D.; Pietka, T.A.; Peche, V.S.; Willecke, F.; Fang, X.; Yu, S.Q.; Scerbo, D.; Chang, H.R.; et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Investig. 2018, 128, 4329–4342.
- Greenwalt, D.E.; Scheck, S.H.; Rhinehart-Jones, T. Heart CD36 expression is increased in murine models of diabetes and in mice fed a high fat diet. J. Clin. Investig. 1995, 96, 1382–1388.
- Iso, T.; Maeda, K.; Hanaoka, H.; Suga, T.; Goto, K.; Syamsunarno, M.R.; Hishiki, T.; Nagahata, Y.; Matsui, H.; Arai, M.; et al. Capillary endothelial fatty acid binding proteins 4 and 5 play a critical role in fatty acid uptake in heart and skeletal muscle. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2549–2557.
- Elmasri, H.; Karaaslan, C.; Teper, Y.; Ghelfi, E.; Weng, M.; Ince, T.A.; Kozakewich, H.; Bischoff, J.; Cataltepe, S. Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3865–3873.
- Masouye, I.; Hagens, G.; Van Kuppevelt, T.H.; Madsen, P.; Saurat, J.H.; Veerkamp, J.H.; Pepper, M.S.; Siegenthaler, G. Endothelial cells of the human microvasculature express epidermal fatty acid-binding protein. Circ. Res. 1997, 81, 297–303.
- Hagberg, C.E.; Falkevall, A.; Wang, X.; Larsson, E.; Huusko, J.; Nilsson, I.; van Meeteren, L.A.; Samen, E.; Lu, L.; Vanwildemeersch, M.; et al. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 2010, 464, 917–921.
- Van der Vusse, G.J. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307.
- Fung, K.Y.Y.; Fairn, G.D.; Lee, W.L. Transcellular vesicular transport in epithelial and endothelial cells: Challenges and opportunities. Traffic 2018, 19, 5–18.
- Minshall, R.D.; Sessa, W.C.; Stan, R.V.; Anderson, R.G.; Malik, A.B. Caveolin regulation of endothelial function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L1179–L1183.
- Glatz, J.F.; Nabben, M.; Heather, L.C.; Bonen, A.; Luiken, J.J. Regulation of the subcellular trafficking of CD36, a major determinant of cardiac fatty acid utilization. Biochim. Biophys. Acta 2016, 1860, 1461–1471.
- Hasan, S.S.; Fischer, A. The Endothelium: An Active Regulator of Lipid and Glucose Homeostasis. Trends Cell Biol. 2021, 31, 37–49.
- Faulkner, A. Trans-endothelial trafficking of metabolic substrates and its importance in cardio-metabolic disease. Biochem. Soc. Trans. 2021, 49, 507–517.
- Abumrad, N.A.; Cabodevilla, A.G.; Samovski, D.; Pietka, T.; Basu, D.; Goldberg, I.J. Endothelial Cell Receptors in Tissue Lipid Uptake and Metabolism. Circ. Res. 2021, 128, 433–450.
- Pi, X.; Xie, L.; Patterson, C. Emerging Roles of Vascular Endothelium in Metabolic Homeostasis. Circ. Res. 2018, 123, 477–494.
- Goto, K.; Iso, T.; Hanaoka, H.; Yamaguchi, A.; Suga, T.; Hattori, A.; Irie, Y.; Shinagawa, Y.; Matsui, H.; Syamsunarno, M.R.; et al. Peroxisome proliferator-activated receptor-gamma in capillary endothelia promotes fatty acid uptake by heart during long-term fasting. J. Am. Heart Assoc. 2013, 2, e004861.
- Kanda, T.; Brown, J.D.; Orasanu, G.; Vogel, S.; Gonzalez, F.J.; Sartoretto, J.; Michel, T.; Plutzky, J. PPARgamma in the endothelium regulates metabolic responses to high-fat diet in mice. J. Clin. Investig. 2009, 119, 110–124.
- Davies, B.S.; Waki, H.; Beigneux, A.P.; Farber, E.; Weinstein, M.M.; Wilpitz, D.C.; Tai, L.J.; Evans, R.M.; Fong, L.G.; Tontonoz, P.; et al. The expression of GPIHBP1, an endothelial cell binding site for lipoprotein lipase and chylomicrons, is induced by peroxisome proliferator-activated receptor-gamma. Mol. Endocrinol. 2008, 22, 2496–2504.
- Coppiello, G.; Collantes, M.; Sirerol-Piquer, M.S.; Vandenwijngaert, S.; Schoors, S.; Swinnen, M.; Vandersmissen, I.; Herijgers, P.; Topal, B.; van Loon, J.; et al. Meox2/Tcf15 heterodimers program the heart capillary endothelium for cardiac fatty acid uptake. Circulation 2015, 131, 815–826.
- Jabs, M.; Rose, A.J.; Lehmann, L.H.; Taylor, J.; Moll, I.; Sijmonsma, T.P.; Herberich, S.E.; Sauer, S.W.; Poschet, G.; Federico, G.; et al. Inhibition of Endothelial Notch Signaling Impairs Fatty Acid Transport and Leads to Metabolic and Vascular Remodeling of the Adult Heart. Circulation 2018, 137, 2592–2608.
- Harjes, U.; Bridges, E.; McIntyre, A.; Fielding, B.A.; Harris, A.L. Fatty acid-binding protein 4, a point of convergence for angiogenic and metabolic signaling pathways in endothelial cells. J. Biol. Chem. 2014, 289, 23168–23176.
- Hwangbo, C.; Wu, J.; Papangeli, I.; Adachi, T.; Sharma, B.; Park, S.; Zhao, L.; Ju, H.; Go, G.W.; Cui, G.; et al. Endothelial APLNR regulates tissue fatty acid uptake and is essential for apelin’s glucose-lowering effects. Sci. Transl. Med. 2017, 9.
- Bae, H.; Hong, K.Y.; Lee, C.K.; Jang, C.; Lee, S.J.; Choe, K.; Offermanns, S.; He, Y.; Lee, H.J.; Koh, G.Y. Angiopoietin-2-integrin alpha5beta1 signaling enhances vascular fatty acid transport and prevents ectopic lipid-induced insulin resistance. Nat. Commun. 2020, 11, 2980.
- Jang, C.; Oh, S.F.; Wada, S.; Rowe, G.C.; Liu, L.; Chan, M.C.; Rhee, J.; Hoshino, A.; Kim, B.; Ibrahim, A.; et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat. Med. 2016, 22, 421–426.
- Rowe, G.C.; Jiang, A.; Arany, Z. PGC-1 coactivators in cardiac development and disease. Circ. Res. 2010, 107, 825–838.
- Finck, B.N. The PPAR regulatory system in cardiac physiology and disease. Cardiovasc. Res. 2007, 73, 269–277.
- Huss, J.M.; Kelly, D.P. Nuclear receptor signaling and cardiac energetics. Circ. Res. 2004, 95, 568–578.
- Augustus, A.S.; Buchanan, J.; Park, T.S.; Hirata, K.; Noh, H.L.; Sun, J.; Homma, S.; D’Armiento, J.; Abel, E.D.; Goldberg, I.J. Loss of lipoprotein lipase-derived fatty acids leads to increased cardiac glucose metabolism and heart dysfunction. J. Biol. Chem. 2006, 281, 8716–8723.
- Noh, H.L.; Okajima, K.; Molkentin, J.D.; Homma, S.; Goldberg, I.J. Acute lipoprotein lipase deletion in adult mice leads to dyslipidemia and cardiac dysfunction. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E755–E760.
- Nakatani, K.; Masuda, D.; Kobayashi, T.; Sairyo, M.; Zhu, Y.; Okada, T.; Naito, A.T.; Ohama, T.; Koseki, M.; Oka, T.; et al. Pressure Overload Impairs Cardiac Function in Long-Chain Fatty Acid Transporter CD36-Knockout Mice. Int. Heart J. 2019, 60, 159167.
- Umbarawan, Y.; Syamsunarno, M.; Koitabashi, N.; Obinata, H.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Hayakawa, N.; Sano, M.; Sunaga, H.; et al. Myocardial fatty acid uptake through CD36 is indispensable for sufficient bioenergetic metabolism to prevent progression of pressure overload-induced heart failure. Sci. Rep. 2018, 8, 12035.
- Nakatani, K.; Watabe, T.; Masuda, D.; Imaizumi, M.; Shimosegawa, E.; Kobayashi, T.; Sairyo, M.; Zhu, Y.; Okada, T.; Kawase, R.; et al. Myocardial energy provision is preserved by increased utilization of glucose and ketone bodies in CD36 knockout mice. Metab. Clin. Exp. 2015, 64, 1165–1174.
- Koonen, D.P.; Febbraio, M.; Bonnet, S.; Nagendran, J.; Young, M.E.; Michelakis, E.D.; Dyck, J.R. CD36 expression contributes to age-induced cardiomyopathy in mice. Circulation 2007, 116, 2139–2147.
- Umbarawan, Y.; Syamsunarno, M.; Koitabashi, N.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Nagahata-Naito, Y.; Obinata, H.; Sano, M.; Sunaga, H.; et al. Glucose is preferentially utilized for biomass synthesis in pressure-overloaded hearts: Evidence from fatty acid-binding protein-4 and -5 knockout mice. Cardiovasc. Res. 2018, 114, 1132–1144.
- Schaap, F.G.; Binas, B.; Danneberg, H.; van der Vusse, G.J.; Glatz, J.F. Impaired long-chain fatty acid utilization by cardiac myocytes isolated from mice lacking the heart-type fatty acid binding protein gene. Circ. Res. 1999, 85, 329–337.
- Binas, B.; Danneberg, H.; McWhir, J.; Mullins, L.; Clark, A.J. Requirement for the heart-type fatty acid binding protein in cardiac fatty acid utilization. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1999, 13, 805–812.
- Sung, M.M.; Byrne, N.J.; Kim, T.T.; Levasseur, J.; Masson, G.; Boisvenue, J.J.; Febbraio, M.; Dyck, J.R. Cardiomyocyte-specific ablation of CD36 accelerates the progression from compensated cardiac hypertrophy to heart failure. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H552–H560.
- Nagendran, J.; Pulinilkunnil, T.; Kienesberger, P.C.; Sung, M.M.; Fung, D.; Febbraio, M.; Dyck, J.R. Cardiomyocyte-specific ablation of CD36 improves post-ischemic functional recovery. J. Mol. Cell. Cardiol. 2013, 63, 180–188.
- Wysocka, M.B.; Pietraszek-Gremplewicz, K.; Nowak, D. The Role of Apelin in Cardiovascular Diseases, Obesity and Cancer. Front. Physiol. 2018, 9, 557.
- Taegtmeyer, H.; Young, M.E.; Lopaschuk, G.D.; Abel, E.D.; Brunengraber, H.; Darley-Usmar, V.; Des Rosiers, C.; Gerszten, R.; Glatz, J.F.; Griffin, J.L.; et al. Assessing Cardiac Metabolism: A Scientific Statement from the American Heart Association. Circ. Res. 2016, 118, 1659–1701.
- Hill, B.G. A metabocentric view of cardiac remodeling. Curr. Opin. Physiol. 2019, 10, 43–48.
- Giles, A.V.; Sun, J.; Femnou, A.N.; Kuzmiak-Glancy, S.; Taylor, J.L.; Covian, R.; Murphy, E.; Balaban, R.S. Paradoxical arteriole constriction compromises cytosolic and mitochondrial oxygen delivery in the isolated saline-perfused heart. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1791–H1804.
- Schenkman, K.A.; Beard, D.A.; Ciesielski, W.A.; Feigl, E.O. Comparison of buffer and red blood cell perfusion of guinea pig heart oxygenation. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1819–H1825.
- Spiekerkoetter, U.; Wood, P.A. Mitochondrial fatty acid oxidation disorders: Pathophysiological studies in mouse models. J. Inherit. Metab. Dis 2010, 33, 539–546.
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477.
- Gibala, M.J.; Young, M.E.; Taegtmeyer, H. Anaplerosis of the citric acid cycle: Role in energy metabolism of heart and skeletal muscle. Acta Physiol. Scand. 2000, 168, 657–665.
- Opie, L.H. Heart Physiology, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004; pp. 460–484.
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591.
- Umbarawan, Y.; Kawakami, R.; Syamsunarno, M.; Koitabashi, N.; Obinata, H.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Hayakawa, N.; Sunaga, H.; et al. Reduced fatty acid uptake aggravates cardiac contractile dysfunction in streptozotocin-induced diabetic cardiomyopathy. Sci. Rep. 2020, 10, 20809.