Streptococcus suis is a pathogen of pigs that can cause infections in humans who are in close contact with infected animals and/or contaminated pork-derived products, as well as those who have consumed raw pork products. Several molecular methods have been applied to investigate S. suis strain diversity and identify phylogenetic groups. Multilocus sequence typing (MLST), commonly used to differentiate between S. suis strains, has been instrumental in identifying that the species is genetically highly diverse. Recent advances in whole-genome analysis have resulted in schemes permitting the classification of S. suis populations as pathogenic or non-pathogenic, or disease-associated or non-disease associated.
1. Introduction
Streptococcus suis is a pathogen of pigs that can cause infections in humans who are in close contact with infected animals and/or contaminated pork-derived products, as well as those who have consumed raw pork products [
1].
S. suis can cause meningitis, septicemia, endocarditis, and arthritis in humans [
2,
3,
4]. Human
S. suis cases have been reported in most countries of Western Europe, North and South America, Africa, Australia, New Zealand, India, Japan, and several other East and Southeast Asian countries, particularly China, Vietnam, and Thailand [
1,
3,
4]. Indeed,
S. suis has been shown to be responsible for thousands of human cases, of which 90.2% were in Asia, 8.5% in Europe, and 1.3% in other parts of the world [
4].
Previously,
S. suis had been classified into 35 serotypes (serotype 1/2, and 1–34) and which were then reduced to 33 serotypes because serotypes 32 and 34 were re-classified as
S. orisratti [
5,
6,
7,
8,
9]. In 2013,
S. suis serotypes 20, 22, 26, and 33 were proposed to be removed from the
S. suis taxon [
10]: serotypes 20, 22, and 26 were proposed as
Streptococcus parasuis [
11,
12], while serotype 33 was classified as
Streptococcus ruminantium [
13]. Hence, currently there are 29 true
S. suis serotypes. Of them, serotype 2 is the most prevalent in both human and pig infections although cases caused by serotypes 4, 5, 9, 14, 16, 21, 24, and 31 have also been reported [
1,
2,
3,
4,
14,
15,
16,
17].
Studying the population structure and the genetic diversity of S. suis is helpful to understand the epidemiology of this organism as well as reveal clones or clonal groups with an apparently increased capacity to cause disease, or which are potentially associated with particular clinical manifestations. Several molecular typing techniques have been applied to the study of S. suis genetic diversity (Table 1).
Table 1. Characteristics of the molecular epidemiological methods for the Streptococcus suis study.
|
Characteristic
|
WGS
|
MLST
|
Multiplex PCR-CC
|
RAPD
|
PCR-RFLP
|
MLVA
|
AFLP
|
PFGE
|
Ribotyping
|
|
Reproducibility
|
Good
|
Good
|
Good
|
Poor to moderate
|
Moderate
|
Good
|
Good
|
Good
|
Good
|
|
Discriminatory power
|
Excellent
|
High
|
Moderate
|
Moderate to good
|
Poor to moderate
|
Excellent
|
Excellent
|
Excellent
|
Good
|
|
Ease of use
|
Moderately labor-intensive
|
Simple to moderate labor
|
Simple
|
Simple
|
Simple
|
Simple
|
Moderate
|
Labor-intensive
|
Labor-intensive
|
|
Interpretation
|
Moderate to very complex
|
Simple to moderate
|
Simple
|
Moderate to complex
|
Simple
|
Simple
|
Complex
|
Moderate to complex
|
Moderate to complex
|
|
Cost
|
Very high
|
Moderate
|
Low
|
Low
|
Low
|
Low to moderate
|
Moderate
|
High
|
High
|
|
Universal applicability
|
Yes
|
Yes
|
Limit to some CCs
|
Yes
|
Yes
|
Yes
|
Yes
|
Yes
|
Yes
|
2. Multilocus Sequence Typing (MLST)
MLST is considered the gold standard to determine the structure of
S. suis populations. This powerful method uses genetic variation that accumulates very slowly in housekeeping genes to investigate the genetic diversity of
S. suis and its use has allowed global and long-term epidemiology. King et al. established the MLST scheme for
S. suis in 2002, using seven different house-keeping genes (
cpn60,
dpr,
recA,
aroA,
thrA,
gki, and
mutS) [
26]. Since its establishment, this MLST scheme has been adopted by multiple laboratories throughout the world and used to determine the STs of
S. suis strains isolated from pig and human cases of infection [
1].
Thus far (28 November 2019), a total of 1245 STs have been recorded in the
S. suis MLST database (
https://pubmlst.org/ssuis/). This method has revealed the presence of many clonal complexes (CCs) within the
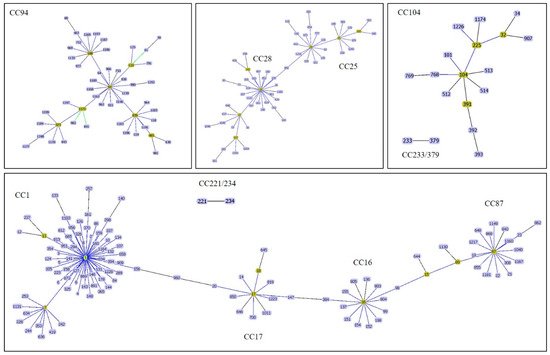
S. suis population in the database. Among the different CCs identified, the most important causes of infections in human and pig have been CC1, CC16, CC20, CC25, CC28, CC94, CC104, CC233/379, and CC221/234 (
Figure 1) [
1,
4,
17,
33]. Different CCs were found to be distributed in different regions of the world [
1]. CC1 was found mostly in Europe, Asia (Cambodia, China, Korea, Japan, Thailand, and Vietnam), Australia, and South America (Argentina), while CC20 (ST20) was described as being important in the Netherlands [
1,
33,
34,
35,
36,
37]. Furthermore, ST7 (CC1), responsible for the 1998 and 2005 epidemics, was mostly present in China [
1] and CC16 and CC94 were predominant in Europe, although human cases were reported in Thailand [
17,
33]. CC25 or CC28 were reported in North America and were also recovered in Thailand, Korea, Japan, and Australia [
1,
4,
33,
34,
35,
36,
37]. Finally, CC104 (ST101, ST104, ST391-ST393, ST512-ST514), and CC233/379 (ST233 and ST379) were endemic to Thailand [
1,
33]. In North America, the structure of
S. suis strains is more complicated and pathotypes are different from Europe and Asia [
38]. Serotypes 1, 1/2, 2, 7, 14, and 23 as well as ST1, ST13, ST28, ST94, ST108, ST961, and ST977 have recently been described as pathogenic strains in the USA [
38].
Figure 1. A goeBURST analysis of major clonal complexes of S. suis causes of infections in human and pig. CC1 is related to CC17 whereas CC17 is closely related to CC16. CC16 and CC87 are related via ST15 and ST89. CC25 and CC28 are related via ST856. Other CCs are independent.
An alternative MLST approach using matrix-assisted laser desorption ionization-time of flight mass spectrometry (MS-MLST) has been reported to be more rapid in providing typing results than dideoxy sequencing [
36]. While both MS-MLST and conventional MLST had 100% concordance in their classification of sequence types (STs), in addition to faster time-to-results, MS-MLST had lower labor requirements and per-isolate costs. MS-MLST analysis was easier to instrument when large numbers of isolates were involved [
36]. However, MS-MLST requires a high initial investment in the MALDI-TOF MS equipment and software. Therefore, this approach may not be readily available to most laboratories, particularly those in developing countries.
3. Prediction of CC Using Polymerase Chain Reaction
Although informative, MLST is a high-cost and time-consuming method that is not ideal for screening large numbers of isolates. MLST is also unavailable in many laboratories in developing countries. Therefore, polymerase chain reaction (PCR)-based approaches [
38,
39,
40,
41,
42] to identify the most important
S. suis CCs (PCR-CC) were developed allowing for the rapid screening of a large number of isolates at a relatively modest cost (summarized in
Table 2).
Table 2. PCR-predicted important clonal complexes of S. suis isolates.
|
Clonal Complexes
|
PCR Methods
|
|
Multiplex PCR
|
PCR of ofs Genes
|
PCR-Pilus-Associated Gene Profiles
|
RAPD
|
16S-23S rDNA PCR-RFLP
|
|
CC1
|
Yes
|
Yes
|
Yes
|
Yes
|
Yes
|
|
CC16
|
No
|
No
|
No
|
Yes
|
Yes
|
|
CC20 **
|
No
|
No
|
No
|
No
|
No
|
|
CC25
|
Yes
|
Yes *
|
Yes *
|
Yes
|
Yes *
|
|
CC28
|
Yes
|
Yes *
|
Yes *
|
Yes
|
Yes *
|
|
CC94
|
No
|
Yes *
|
No
|
No
|
No
|
|
CC104
|
Yes
|
Yes *
|
Yes
|
Yes
|
Yes *
|
|
CC233/379
|
Yes
|
No
|
No
|
Yes
|
Yes *
|
|
CC221/234
|
Yes
|
No
|
No
|
Yes
|
Yes
|
Note: * reveal the same profile; thus, could not be differentiated for each. ** PCR methods were not applied to CC20.
4. Whole-Genome Sequencing Approaches
WGS approaches have increasingly been used to investigate
S. suis isolates, including molecular determination of serotype [
44], characterization of outbreaks [
45,
46], evaluation of
S. suis reinfection [
47], and to determine the population structure of
S. suis isolates of serotype 2 belonging to ST25 and ST28 [
48,
49], as well as of serotype 9 [
50]. WGS-based bacterial typing strategies commonly use one of two approaches: SNP (sequence)-based, or MLST (allele)-based. The SNP approach compares single nucleotide differences between isolates in comparison to a reference genome and is particularly useful to determine the clonal relationship between highly similar isolates. The MLST approach is an extension of conventional 7-gene MLST that expands the range of genes to the genome scale and can be roughly divided into core-genome-based MLST (cgMLST) and whole-genome-based MLST (wgMLST). Allele-based comparisons can be conducted using analysis tools available on the internet, such as BacWGSTdb (
http://bacdb.org/BacWGSTdb/) [
51], pubMLST (
https://pubmlst.org/ssuis/), or the Center for Genomic Epidemiology (
https://cge.cbs.dtu.dk/services/MLST/) [
52].
The definition of virulence in
S. suis remains controversial, and WGS-based approaches have played an important role in the efforts to identify virulence markers permitting to differentiate between commensal organisms and those prone to cause disease. A comparative genome hybridization (CGH) was used to analyze 55
S. suis isolates from different serotypes, recovered from different hosts, different clinical sources, and different geographical locations [
53]. Clustering of CGH data divided
S. suis isolates into 2 clusters. Cluster A exclusively contained virulent CC1 isolates of serotypes 1 and 2 isolates [
53]. Cluster B, on the other hand, contained mainly a more divergent and heterogeneous group of serotype 7 and 9 isolates [
53]. Another study used CGH on 39 isolates of different serotypes, sources, geographic locations, isolation years, and STs. This study revealed that the
S. suis strains tested could be classified into three groups of differing levels of virulence: (i) epidemic and highly virulent (E/HV group), which included ST1, ST6, ST7, and ST11 isolates; (ii) virulent (V group), containing ST81, ST13, ST56, ST87, ST308, ST54, and ST53 isolates; and (iii) intermediately or weakly virulent (I/WV group), composed of isolates belonging to several STs that were all recovered from non-human sources [
28].
The genomes of 375
S. suis isolates with detailed clinical phenotypes from pigs and humans from the United Kingdom and Vietnam were analyzed using a Bayesian Analysis of Population Structure (BAPS) [
54]. The study showed clear genetic differences between systemic, respiratory, and non-clinical (carriage)
S. suis isolates. Interestingly, systemic isolates had a smaller genome than respiratory and carriage isolates, and they tended to have an over-representation of virulence factors and a larger number of genes involved in defense functions [
54]. Willemse et al. (2016) used BAPS to investigate the whole genomes of 98
S. suis isolates from human patients and pigs with invasive disease in the Netherlands, and 18 genomic complete and available
S. suis sequences. The BAPS-based population grouping did not correlate with serotype but correlated well with the CC of the isolates. BAPS Group 1 comprised all CC13 isolates, BAPS Group 2 comprised most of the CC16 isolates, BAPS Group 4 comprised all CC1 isolates, BAPS Group 5 comprised both the CC27 and the CC29 isolates, and BAPS Group 6 comprised most CC20 isolates [
29].
This entry is adapted from the peer-reviewed paper 10.3390/pathogens9020081