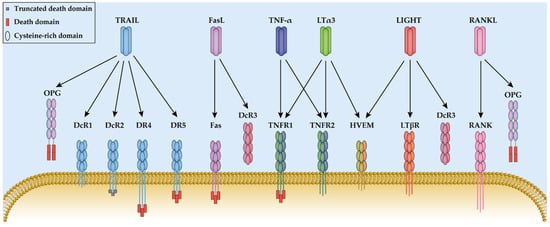
Tumor necrosis factor (TNF or TNF-α) and lymphotoxin (LT) are the first two cytokines that have been characterized as TNF superfamily members. They have a homologous amino acid sequence and were both discovered based on their anti-tumor effects [
8]. Moreover, both ligands bind to TNF receptor type 1 and type 2 (TNFR1 and TNFR2) and mediate similar cell signaling transduction. Thus, it is necessary to understand and compare the role of TNF-α and LT in pathways and diseases in order to discriminate the TNFR1- or TNFR2-specific treatments and their medical effects.
2.1. Tumor Necrosis Factor and Lymphotoxin
TNF-α was first identified in 1975 based on its ability to induce necrosis in transplanted tumors and the absence of toxicity to embryo in mice [
9]. It is mainly expressed by activated macrophages, T lymphocytes, and natural killer (NK) cells [
10]. It is synthesized as a cell surface transmembrane protein with 233 amino acids (26 kDa) forming a stable homotrimer, which contains a TNF family homology domain in the extracellular part, a transmembrane domain, a spacer stalk and an intracellular domain [
11]. The transmembrane TNF-α (mTNF-α) is subsequently proteolytically cleaved on the spacer stalk by TNF-α-converting enzyme (TACE, or ADAM17) resulting in the release of a 17 kDa soluble TNF-α (sTNF-α) [
12]. Both forms are biologically active as homotrimers and capable of binding with TNFR1 and TNFR2, whereas sTNF-α shows a lower affinity to TNFR2 compared with mTNF-α and only activates TNFR1 to mediate cellular signal transduction [
13].
LT, a cytokine secreted by lymphocytes, was first identified in 1968 based on its in vitro anti-tumor activity [
14,
15]. Due to its similar effects of LT-α on tumor cells to TNF-α, it was renamed TNF-β in 1985 [
16]. However, the discovery of the subunit LT-β revealed its additional functions other than TNF-α and led to the reversion of the name to LT. LT-α is the only TNF ligand without a transmembrane domain and is directly secreted into the cell exterior, while LT-β expresses as a transmembrane protein [
17]. Three LT-α subunits form a homotrimer LTα3 that binds to TNFR1, TNFR2, and the herpes virus entry mediator (HVEM). LT-α and LT-β can also assemble the cell surface-bound heterotrimer LTα1β2, which is the predominant form that binds with the lymphotoxin β receptor (LTβR), and the LTα2β1, which binds to TNFR1 and TNFR2 with a minor physiological effect [
17,
18]. In this section, the LT-related TNFR1 and TNFR2 pathways are discussed, while the HVEM and LTβR pathways are discussed in the LIGHT section.
It has been demonstrated that TNF-α and LTα3 have similar affinities to TNFR1 and are able to transduce downstream signaling in a similar way [
19]. Interestingly, sTNF-α cannot initiate TNFR2, but LTα3, as a soluble form, is able to transduce signaling through TNFR2 [
20]. As they have similar amino acid sequences in their TNF homology domain (THD), their different function may be due to differences in their tertiary structure within subunits or the modular assembly mode in trimer formation.
TNF-α and LTα3 both bind to TNFR1 or TNFR2 to initiate similar signaling transduction; therefore, we will mainly focus on TNF-α-related research with a tacit understanding that LTα3 shows comparable performance. TNFR1 and TNFR2 mediate common and exclusive pathways to induce cell proliferation, inflammation, apoptosis, or necroptosis [
21]. Thus, it is important to understand the different roles of TNFR1 and TNFR2 in cell signal transduction.
2.2. The TNFR1 and TNFR2 Induced Signal Transduction
TNFR1 protein expression is detected in almost all human nucleated cells, whereas TNFR2 is mainly expressed in the immune system and endothelial tissue [
22], such as macrophages [
23], T cells [
24], monocytes [
25], endothelial progenitor cells [
26], and mesenchymal stem cells [
27]. Both TNFR1 and TNFR2 are transmembrane glycoproteins with four N-terminal cysteine-rich domains (CRDs) in their extracellular domain and a transmembrane domain [
28]. Both receptors are capable of activating the nuclear factor kappa B (NF-κB) and activator protein-1 (AP-1) pathway, but TNFR1 also has the capacity to induce cell necroptosis and apoptosis due to the presence of the death domain (DD) [
29]. TNF-α/LTα3 binds to TNFR1 to recruit TNFR1-associated death domain protein (TRADD), the receptor-interacting protein 1 (RIP1), the TNF receptor-associated actor 2 (TRAF2) and the cellular inhibitor of apoptosis proteins 1/2 (cIAP1/2). For TNFR2, only TRAF2 and cIAP1/2 are recruited. The complex formation initiates the downstream cascades and ultimately translocates NF-κB and AP-1 to the nucleus to trigger their downstream gene expression, such as FLICE-like inhibitory protein (FLIP). Occasionally, when there is insufficient FLIP, the internalized TNF-α/TNFR1 complex recruits Fas-associated death domain protein (FADD) and procaspase-8 to form the death-inducing signaling complex (DISC) and generate apoptosis signaling [
30,
31]. Under other circumstances, the deficiency of active caspase-8 results in the binding of RIP3 to RIP1 to assemble necroptosome and induce necroptosis [
32].
2.3. Engineering TNFR1-Specific Ligands
Due to the different binding affinities between TNF-α and its two receptors, as well as the diverse pathways they trigger, various attempts at specifically targeting the TNFR1 receptor have been made to achieve precise drug delivery, increase effectivity, and reduce TNF-α-related inflammation (
Table 1). The research was initially focusing on the construction of mutated TNF-α to target either TNFR1 or TNFR2. In 1993, Ostade et al. constructed two TNF-α muteins,
L29S and
R32W, which are specific to TNFR1 [
33]. Research showed that even though the artificial R32W had lower affinity to both receptors, it still showed equivalent cytotoxicity to colon and laryngeal cancer cell lines compared with wildtype (WT) TNF-α [
33,
34]. Then, to regain the full binding capacity of R32W to TNFR1, a double mutant
R32W-S86T was created, with none binding to TNFR2 [
35]. R32W-S86T was widely analyzed and proved to be lethal to tumor cells with low pro-inflammatory effects [
34]. Nevertheless, these aforesaid mutants have similar shortages as observed for WT TNF-α, which cause coronary vasoconstriction, hypotension, anti-lipogenesis in vivo, and a short half-life [
36,
37]. Thus, more efforts were made to solve these problems. By introducing triple site mutations in position 5, 6, and 29 in TNF-α, Atarashi et al. created a mutein,
F4614, which exhibited higher anti-tumor effects and reduced hypotensive risk more effectively than WT TNF-α [
38,
39]. Another TNFR1-specific mutein,
M3S, was made to obtain a higher thermal stability, two-fold prolonged half-life and lower systematic cytotoxicity in vivo. However, the multiple site mutations also resulted in a lower binding affinity to TNFR1 and the M3S did not perform well in animal assays [
40]. Recently, a
mutTNF G4 with mutations from sites 84 to 89 was identified, showing higher affinity to TNFR1 than WT TNF-α [
41]. The intravenous injection of mutTNF G4 induces the permeabilization of the blood–brain barrier (BBB), which successfully enhances the delivery of the therapeutic reagent into brain tumor.
Table 1. The mutation sites and binding affinity of the receptor-specific TNF-α variants.
|
Ligand
|
Specificity
|
Variants
|
Mutation Sites
|
Binding Affinity (nM)
|
Ref.
|
|
TNFR1
|
TNFR2
|
|
TNF-α
|
TNFR1 &TNFR2
|
WT
|
/
|
15.8
|
35.3
|
[41]
|
|
TNFR1
|
L29S
|
L29S
|
− 1
|
−
|
[42]
|
|
R32W
|
R32W
|
−
|
−
|
[42]
|
|
R32W-S86T
|
R32W/S86T
|
3540
|
NB 2
|
[42]
|
|
F4614
|
T5G/P6D/R29V
|
−
|
−
|
[38]
|
|
M3S
|
L29S/S52I/Y56F
and 449/455del
|
−
|
−
|
[40]
|
|
mutTNF G4
|
A84S/V85S/S86T/
Q88N/T89P
|
8.72
|
NB
|
[41]
|
|
TNFR2
|
D143N-A145R
|
D143N/A145R
|
NB 2
|
13.1
|
[42]
|
1–means not available. 2 NB means no binding.
Investigations of TNF-α antibodies, including both antagonist and agonist antibodies, have been carried out for decades. The antagonist antibody binds to the target protein and blocks its immune responses; conversely, the agonist antibody activates the checkpoint protein and initiates the signaling transduction [
43]. FDA-approved antibodies are now commercially available, including infliximab, adalimumab, certolizumab pegol, and golimumab, which are all TNF-α antagonist antibodies [
44]. However, the complete blockage of the TNF-α pathway leads to poor results with fatal side effects and low resistance to infections [
45]. Therefore, apart from engineering TNF-α variants, the antagonistic antibody specifically targeting TNFR1 attracted more attention. To understand the receptor-specific functions in mice model,
HS1097 and
DMS5540 targeting and inhibiting TNFR1 were made and commercialized [
46,
47]. In mice model, the application of HS1097 reduces experimental autoimmune encephalomyelitis symptoms, and DMS5540 effectively suppresses collagen-induced arthritis (CIA) without additional effector T cell activation and inflammation reaction [
48,
49]. Further, a mouse TNFR1 antibody was humanized and named
ATROSAB. It has a similar affinity to both rhesus and human TNFR1, which makes it a potential agent for the preclinical trials in the CIA model of rhesus monkeys [
50]. ATROSAB binds to TNFR1 without signal activation and blocks the activity of both TNF-α and LTα3, leaving the TNFR2 pathway intact [
51].
In addition to antibodies, small molecules that inhibit specific receptors are gaining popularity due to their low cost and convenient drug administration. Chen et al. utilized pharmacophore model filtering and molecular docking to obtain 10 virtual hits and evaluated their binding affinity to TNF-α and TNFR1 [
52]. Three compounds out of ten suppress TNF-α induced cytotoxicity in the noncancerous cell line L929 in a dose-dependent manner. Nevertheless, all the selective compounds inhibit both TNF-α and TNFR1 and thus are lacking receptor specificity. Based on the crystal structure of TNF-α-TNFR1 complex, the TNF-α inhibitor
ZINC09609430, the TNFR1 inhibitor
ZINC02968981, and the TNF-α–TNFR1 complex inhibitor
ZINC05462670 were selected [
53]. However, the mentioned compounds only underwent simulation prediction; therefore, more biological experiments need to be done to confirm their efficiency. Another strategy was employed to retrieve seven promising compounds targeting TNFR1 by the high-throughput screening of the ChemBridge DIVERSet library. They proved that the selected noncompetitive inhibitors
DS41 and
DSA114 significantly block TNF-α/LTα3-induced NF-κB activation in a TNFR1-specific pattern through perturbing the conformation of TNFR1 without interfering in ligand-receptor assembly [
54]. Compared with the development of TNFR1-specific mutants or antibodies, the small molecule inhibitors are still in the early progression stage.
2.4. TNFR2-Specific Applications
TNFR2 has different features to TNFR1 because of its absence of a DD in the intracellular part. The TNFR2-specific mutant protein was mainly used to investigate the receptor-specific signaling pathways and pathogenesis (
Table 1), such as, the
D143N-A145R with extremely low binding to TNFR1 [
35]. The mutation sites were selected based on a library of site-directed mutants of human TNF-α, and it was shown that sites 143–145 are responsible for binding with TNFR1 but not with TNFR2. This mutein is frequently used in a comparison with R32W-S86T to explore the functions of different receptors [
55,
56,
57]. Even though these recombinant proteins were made to target TNFR2, more investigations of TNF-α and its receptors have indicated that TNFR2 is incapable of being activated by binding with sTNF-α.
In contrast to TNFR1, TNFR2 is crucial for the generation and proper functioning of regulatory T cells (Tregs). It regulates immune suppression and promotes apoptosis of autoreactive T cells in multiple diseases [
58,
59]. Due to the key role of TNFR2 in the immune system, researchers have attempted to develop specific TNFR2 agonists by mimicking mTNF-α. Therefore, based on the achieved TNFR2-specific mutated TNF-α, it was fused with other protein domains to form spontaneous oligomer and to increase the avidity. The fusion protein
STAR2 is constructed by fusing a mutated single-chain mouse TNF-α trimer (D221N-A223R) with one domain of chicken tenascin C (TNC) and three of the fused complexes were trimerized to form a nonameric molecule [
60]. STAR2 induced the expansion of Tregs without pro-inflammatory side effects [
60]. However, it also showed a mixed result in myocardial infarction mice, which improved the left ventricular function yet decreased the survival rate [
47]. In addition, a human TNFR2-specific fusion protein (
TNC-scTNFR2) was created with a single-chain human TNF-α mutant (D143N-A145R) and a human TNC instead of a chicken derivative, which yielded a similar 3D structure to STAR2. It established a neuroprotective effect through preventing oxidative stress-induced cell death from H
2O
2 and catecholaminergic in human dopaminergic neuronal cell line LUHMES [
61]. Along with using TNC, the dimerization domain EHD2 derived from the heavy chain domain CH2 of IgE was also fused with human TNF-α (D143N-A145R) to form a dimer (
EHD2-scTNFR2) under nonreducing conditions. It was shown that EHD2-scTNF
R2 protects mice from acute neurodegeneration and memory impairment, and the murine ortholog EHD2-sc-mTNF
R2 induces the expansion of Tregs with anti-inflammatory responses [
58,
59]. Unlike TNFR1-specific application, the research into TNFR2 related treatments is still in the early phase. With greater recognition of the crucial role of TNFR2 in neuroprotection and immune system counterpoise, more research is expected to be seen in the future.