Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Acetylation, a reversible epigenetic process, is implicated in many critical cellular regulatory systems including transcriptional regulation, protein structure, activity, stability, and localization. Lysine acetylation is the most prevalent and intensively investigated among the diverse acetylation forms.
- lysine acetylation
- cancer hallmarks
- cold atmospheric plasma
- onco-therapeutics
1. Lysine Acetylation Classification and Basic Features
Over 80% of human proteins bear N-terminal acetylation at the α-amino position of the first amino acid [12]. Acetylation of key lysine residues can occur enzymatically or non-enzymatically to affect its intermolecular interactions, functionalities, stability, and localization. Lysine acetylation had once been considered as a unique modification of histones but has recently been found residing in thousands of non-histone proteins located in almost every cellular compartment such as nucleus, mitochondria, and cytoplasm.
Lysine acetylation is a reversible epigenetic modification controlled by lysine acetyltransferases (KATs) and lysine deacetylases (KDACs, Sirtuin). As such, it represents an important epigenetic modulatory process of both histone and non-histone proteins. During lysine acetylation, an acetyl group is transferred from acetyl-coenzyme A (acetyl-CoA) to the ε-position of the lysine side chain of a protein, where the positive electrostatic charge of that specific loci is neutralized. Non-enzymatic acetylation also occurs, the level of which is determined by the counteracting effects of the ‘writer’ (acetyltransferases) and the ‘eraser’ (deacetylases). Acetylated lysine residues can be identified by the ‘reader’ proteins that harbor specific acetyl-lysine binding domains for function interpretation.
KATs from distinct families harbor homologous acetyl-CoA binding domains, the flanking regions of which determine their enzyme specificities [13]. The best characterized acetyl-CoA binding domain is the bromodomain. Almost all bromodomain-containing proteins are located in the nucleus to regulate chromatin structure and functions. Other acetyl-CoA binding domains include the PHD domain in proteins DPF3b, CHD4, and KAT6A, as well as the YEATS domain in YEATS2, ENL, AF9, TFIIF, and GAS41 [14,15]. While the bromodomain, PHD, and YEATS domains read acetylated lysine residues, the SET domain identifies non-acetylated lysine enriched regions of H3, KU70, p53, and FOXO1 [16].
KDACs and Sirtuin (SIRT) are mechanistically and structurally distinct deacetylases, which are classified into four categories. Class I, II, and IV deacetylases are Zn2+-dependent and include KDACs 1-11. While class I members (KDAC1/2/3/8) are localized primarily in the nucleus, class II (KDAC4/5/6/7/9/10) and class IV (KDAC11) deacetylases exist both in the nucleus and cytoplasm (Figure 1). Class III deacetylases comprise SIRTs1–7, which are distributed in the nucleus (SIRsT1/2/6/7), cytoplasm (SIRTs1/2/5), and mitochondria (SIRTs3/5) (Figure 1). Out of the seven Sirtuins in mammals, only SIRTs 1/2/3 present robust lysine deacetylase activity [17].
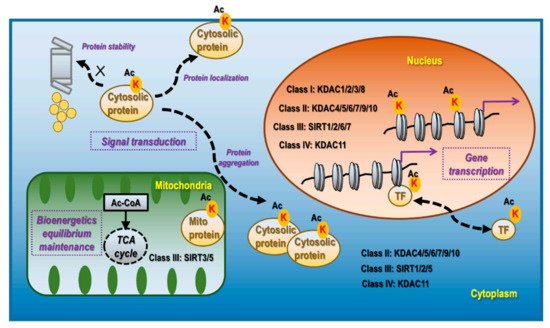
Figure 1. Cellular distribution, corresponding deacetylases, and primary functionalities of lysine acetylation. Lysine acetylation occurs in the nucleus, mitochondria, and cytoplasm. Nucleus lysine acetylation is deacetylated by class I deacetylases KDAC1/2/3/8, class II deacetylases KDAC4/5/6/7/9/10, class III deacetylases SIRT 1/2/6/7, Class IV deacetylase KDAC11; mitochondria lysine acetylation is deacetylated by class III deacetylases SIRT3/5; cytoplasm lysine acetylation is deacetylated by class II deacetylases KDAC4/5/6/7/9/10, class III deacetylase SIRT1/2/5, and class IV deacetylase KDAC11. Nucleus lysine acetylation can affect gene transcription via modulating chromatin structure if occurring on histones and regulate gene expression via controlling the localization, expression, and activity of TFs as well as the transcriptional machinery. Mitochondrial lysine acetylation can control the maintenance of bioenergetic equilibrium as acetyl-CoA is the substrate of acetylation and TCA cycle, and an important product of glycolysis. Cytoplasmic acetylation can affect signal transduction via modulating protein stability, localization, and aggregation, alone or together with other PTM programs.
Acetylation in the nucleus is highly effective in inducing active cell division, including in tumor cells, and can occur on histones [18], transcription factors (TFs) [19], and basal transcription machinery [20]. The acetylation functionalities are associated with gene transcription regulation as summarized in Figure 1. The mechanism of lysine acetylation is related to the exposure of DNA to the transcription machinery by disrupting the electrostatic interactions between the phosphodiester backbones of DNA and lysine enriched nucleosomes [21]. Acetylation modulates transcription factor activities via inducing nuclear translocation, triggering protein stabilization, modifying molecular complex composition, and enhancing the specificity and selectivity of chromatin binding. For instance, acetylation of the TF STAT3 at K685 induces its homodimerization and nucleus translocation [22]; acetylation of the TF p53 at K120 and K382 prevents it from MDM2-mediated ubiquitination and degradation [23]. Besides TFs, many factors associated with the RNA polymerase II complex are acetylated. For example, the C-terminal domain of RNA polymerase II is acetylated in actively transcribed genes, the lack of which leads to polymerase pausing [20].
Acetylation in the mitochondria plays a fundamental role in maintaining the cellular equilibrium of bioenergetics that is enriched in metabolically active tissues such as the liver [24] and the heart [25] (Figure 1). Approximately one-third of mitochondrial proteins are acetylated, many of which harbor multiple acetylated lysines [8]. Acetylated mitochondrial proteins are involved in many functions relevant to cellular metabolism including the TCA cycle, oxidative phosphorylation, lipid β-oxidation, carbohydrate metabolism, nucleotide metabolism, amino acid metabolism, and the urea cycle [26]. For instance, decreased acetylation of two mitochondrial proteins PDHA1 and PDP1 suppresses their functionalities, which leads to altered glucose homeostasis and the Warburg effect [27]; SIRT3 regulates ATP synthase via deacetylating proteins involved in the mitochondrial respiratory chain complexes such as SDHA [28].
Acetylation in the cytoplasm contributes, alone or together with other post-translational modification (PTM) events, to the regulation of signal transduction via affecting protein stability, aggregation, and localization, which are associated with protein turnover, activity, cytoskeleton regulation, and cell migration [29] (Figure 1). Though relatively less studied, it is currently considered to be predominantly present in the liver, peri-renal, testis fat, neuron, and tissues with high levels of acetyl-CoA [17]. For instance, under the context of neuro-degeneration, α-tubulin acetylation at K40, occurring at the luminal side of microtubules, is a marker of protein stability [30]; acetylation of Tau at K280 promotes its aggregation while acetylation at K274 and K281 leads to its mis-localization, and acetylation at K174 slows its cellular turnover, which collectively contribute to cognitive impairment [31]; PD-L1 de-acetylation on K263 in the cytoplasmic domain by HDAC2 is a prerequisite of its nuclear translocation that determines the efficacy of anti-PD-1 immunotherapy [32].
2. Acetylation and Immunity
Immunity, as comprised primarily of adaptive and innate immune responses, corresponds to the cancer hallmarks of ‘avoiding immune destruction’ and ‘tumor-promoting inflammation’, respectively.
As histone acetylation is sustained by acetyl-CoA [47] and metabolic regulation plays central roles in immune responses [60,61], histone acetylation has been implicated in the regulation of both adaptive and innate immunity. Regarding the adaptive immune response, fish oil influences the histone acetylation of neonatal T-cell genes implicated in adaptive immunity in a number of ways, such as (i) increasing H3 acetylation at FOXP3, IL10RA, IL7R, and H4 acetylation at CD14 [62]; (ii) inducing LDHA, an enzyme converting pyruvate and NADH to lactate and NAD+, (iii) promoting INFϒ expression in activated T cells via elevating H3K9 acetylation, a histone mark of active transcription [63]; (iv) ligating glucocorticoid-induced TNFR-related protein (GITR), a costimulatory molecule of effect T cells and regulatory T cells (Tregs), on activated CD4+ T cells, which subverts the induction of FOXP3+ Tregs and directs these CD4+ T cells to Th9 cells towards boosted Th9 adaptive immune responses via FOXP3 deacetylation at H3K9 and H3K27 [64]. Loss of H3K27 acetylation in the enhancer region of CREBBP leads to aberrant transcriptional silencing of genes that regulate B cell signaling and adaptive immune response including class II major histocompatibility complex (MHC) and consequently contribute to lymphomagenesis [65]. Regarding innate immune response involving histone acetylation, lipopolysaccharide (LPS) signaling directly promotes the incorporation of acetyl-CoA into histones, leading to enhanced H4K27 acetylation and induction of genes associated with inflammatory responses such as IL-6, IL-12p40, and IL-12p70 [66]; inhibiting endothelial NOTCH1 signaling reduces cytokine-mediated H3K27 acetylation in a subset of NFκB-directed inflammatory enhancers that weaken endothelial up-regulation of adhesion molecules and recruit less leukocytes to the inflammatory sites [67].
Nonhistone acetylation, if residing in genes regulating core elements of the adaptive or innate immune response, could lead to aberrant immune and inflammatory responses. As examples of the adaptive immune response, pyruvate kinase M2 (PKM2) acetylation at K433 is critical for its de-tetramerization and nuclear translocation, where de-tetramerization helps activate dendritic cells as a result of its decreased enzymatic activity and nuclear localization makes it possible for PKM2 to form a complex with c-Rel towards the enhanced Il12p35 expression and Th1 cell differentiation [68]; STAT3 acetylated at K87 binds BRD2 and is recruited to active enhancers occupied with TFs IRF4 and BATF, potentiating the genetic program required for Th17 cell differentiation and development [69]. Regarding examples on innate immune response, MKP1 acetylation at K57 reduces innate immune signaling and inflammation via blocking the MAPK pathway that plays pivotal roles in toll-like receptor signaling [70]; acetylation of α-tubulin at K40 is involved in augmentation of the NLRP3 inflammasome that enhances the innate immunity of the host [71]. Moreover, lysine acetylation of NKG2D ligand Rae-1 at K80 and K87 sensitizes tumor cells to NKG2D immune surveillance via NKG2D stabilization that involves both adaptive and innate immune responses [72].
3. Combining HDAC Inhibitors with Cold Atmospheric Plasma Provides Novel Onco-Therapeutic Opportunities
HDAC isoenzymes were reported to be high in malignant cells [109]. HDAC inhibitors can reactivate the expression of tumor suppressors and have emerged as one type of well-characterized epigenome-targeting agents that are capable of suppressing both histone acetylation and non-histone acetylation to resolve tumors [110], halt cancer metastasis [111], reprogram cancer cell metabolism [112,113], modulate the immune [114], chemo [115] and radio [116] sensitivity of cancer cells. Several HDAC inhibitors have been developed and approved by the Food and Drug Administration (FDA) for cancer treatment that represent distinct HDAC specificity. For instance, vorinostat (Zolinza; Merck) [117], panobinostat (arydak; Novartis) [118], belinostat (Beleodap; Spectrum Pharmaceuticals) [119], and romidepsin (Istodax; Celgene) [120] have been approved for treating refractory multiple myeloma and cutaneous/peripheral T-cell lymphoma, where the first three drugs target HDAC1/2/3/6 and the last one inhibits HDAC1/2/3 [121].
Like other targeted therapies, cancer cells may also develop resistance to HDAC inhibitors due to, e.g., the use of alternative PTM programs shutting down gene transcription such as DNA methylation [122], the adoption of HSP90-induced unfolded protein response pathway, elevated stress-responsive TF such as NFκB (also named p65), enhanced level of anti-apoptotic proteins such as Bcl-2, and upregulated cellular anti-oxidant signaling [123]. These render it possible to combine HDAC inhibitors with other therapeutic strategies. By reviewing literatures on combinatorial strategies involving the four FDA-approved HDAC inhibitors vorinostat, panobinostat, belinostat, and romidepsin in the past 5 years, we found that inhibitors of kinases and their downstream effectors such as gefitinib, inducers of apoptosis and cell cycle arrest such as riluzole, and autophagy inhibitors such as quinacrine are typically combined with HDAC inhibitors for improved cell death; anti-angiogenetic agents such as bevacizumab and drugs targeting cancer stemness such as minocycline are commonly used together with HDAC inhibitors to halt metastasis; inhibitors of key enzymes involved in cell metabolisms such as simvastatin is primarily combined with HDAC inhibitors to target cancer cell metabolism; anti-inflammation agents such as dexamethasone, and drugs designed for immune-targeting such as rituximab are commonly used together with HDAC inhibitors towards reduced inflammation and enhanced immunotherapeutic efficacy; and alkylating agents such as cisplatin, topoisomerase inhibitors such as cytarabine, DNA synthesis inhibitors such as idarubicin, p53 activators such as CBL0137 are typically used together with HDAC inhibitors to toggle the genome integrity of cancer cells; inhibitors of HSP90 and proteosome such as 17AAG and ixazomib, DNA or histone methylation such as 5-azacytidine and 3-deazaneplanocin A can be jointly used with HDAC inhibitors to target any relevant cancer hallmark (Supplementary Table S1).
Cold atmospheric plasma (CAP), a cocktail of reactive oxygen and nitrogen species generated by ionization of gas via an electromagnetic field, has been proposed as an emerging therapeutic approach [124,125] with numerous studies demonstrating its selectivity against diverse cancer cells [126]. As CAP could selectively trigger apoptosis in many malignant cells [127,128,129], significantly reduce the amount of active NFκB that halts the migration of MDA-MB-231 triple-negative breast cancer (TNBCs) cells [126] and selectively eliminate more aggressive breast cancer cells showing mesenchymal attributes [129], target cancer cell metabolism [130], eliminate immunosuppressive pancreatic stellate cells and induce immunogenic cell death [131], and eradicate cancer stem cells that typically feature high anti-oxidative ability [132], the ability of CAP in targeting various cancer hallmarks is evident [125]. CAP has been considered as a relatively mild promising onco-therapeutic approach with minimal adverse effects on healthy cells [133]. Moreover, CAP is known to cause controlled oxidative stress [45,132] that can alter many cell death programs such as cell cycle progression and apoptosis [134]. To aid in the understanding of CAP as a novel therapeutic, recent progress on its other applications is summarized in Supplementary Table S2.
Importantly, it was demonstrated that CAP could be used to modify the human epigenome, with most reports focusing on DNA methylation, histone methylation, and histone acetylation [135]. Through pyrosequencing and whole-genome methylation microarray analysis, Park et al. reported the effect of CAP in triggering hypomethylation of a specific CpG site of Alu from 23.4 to 20.3% in MDAMB231 TNBC cells [136]. Hou et al. revealed the importance of exposure time in determining the epigenetic effect of CAP on cell signaling cascades through studying A549 small lung cancer cells; it was suggested that a 3 min or longer CAP exposure can possibly affect DNA methylation via altering methyltransferase activity [137]. The levels of many non-coding RNAs were reported to be affected by CAP such as miRNA-19a-3p [138] and ZNRD1-1AS1 lncRNA [139]; moreover, the effects of these non-coding RNAs were attributed to CAP-induced DNA methylation alteration in cancer cells as well. Lee et al. investigated the influence of CAP on histone methylation levels using the H3K4me3 (which is associated with activated gene expression) genome-wide ChIP-sequencing approach and showed that H3K4me3 levels were, in general, lower in CAP treated MCF7 cells as compared with the control [140]. Though relatively little has been reported on the effect of CAP on histone acetylation, CAP-treated human mesoderm-derived stem cells were characterized with the increased HDAC1 activity and decreased acetylated histone-3 levels [141]. On the other hand, synergistic killing effects of A549 non-small cell lung cancer cells were achieved by combining HDAC inhibitors and CAP [142]. These seemingly contradictory effects of CAP on histone acetylation in these two studies can be explained by the significant differences of cell signaling networks at the healthy and malignant states that may potentially justify the use of HDAC inhibitors with CAP for selective targeting of cancer cells.
These demonstrated features of CAP not only make it a potential onco-therapeutic strategy but also an excellent remedy to prevent cancer cells from becoming resistant to HDAC inhibitors, rendering the joint use of HDAC inhibitors and CAP a novel and promising combinatorial strategy for cancer treatment. These possibilities require rigorous experimental investigations, among which whether CAP could function as an HSP90 inhibitor or HDAC inhibitor deserves the most attention.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14020346
This entry is offline, you can click here to edit this entry!