Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pathology
Impaired folate-mediated one-carbon metabolism (FOCM) is associated with many pathologies and developmental abnormalities. FOCM is a metabolic network of interdependent biosynthetic pathways that is known to be compartmentalized in the cytoplasm, mitochondria and nucleus.
- Alzheimer’s disease
- COVID-19
- epigenetics
- homocysteine
- Long COVID
- MTHFR gene mutations
- nutritional B vitamin deficiencies
- PASC
- repurposed drugs
- type 2 diabetes mellitus
1. Introduction
There has been renewed interest in FOCM during the past several years due to recent insights, which indicate dietary inadequacies of micronutrients or genetic polymorphisms. These dietary micronutrient deficiencies include the essential water-soluble vitamins B9 (folate-folic acid), vitamin B12 and vitamin B6. Importantly, these vitamin deficiencies are closely linked to CVD, vascular abnormalities including endothelial activation and dysfunction, myocardial diseases (myocardial infarction and abnormal remodeling in congestive heart failure), hypertension, stroke, LOAD, T2DM, neural tube defects, epigenetic abnormalities and cancer [1].
Hcy is a nonessential (non-proteinogenic) sulfur-containing amino acid and an intermediary metabolic product derived from the demethylated essential amino acid methionine [2]. It is commonly known that plasma concentrations of Hcy are inversely related to plasma concentrations of folate, vitamin B12, and vitamin B6, as well as to the intake of these vitamins [2][3][4][5][6][7]. Further, it is now accepted that HHcy has vasculotoxic effects as well as neurotoxic effects that are associated with neuroinflammation, neurodegeneration, pro-oxidation as well as proatherogenic/prothrombotic effects [2][3][4][5][6][7][8]. HHcy plays an important role in the causation of oxidative stress with excess formation of reactive oxygen nitrogen species (RONS) in the vascular endothelium and multiple organ systems due to autoxidation of Hcy, formation of Hcy mixed disulfides, interactions of Hcy thiolactones and protein homocysteinylation [2][8][9][10][11].
FOCM is known to support multiple physiological processes. These include biosynthesis of purines and thymidylate, amino acid homeostasis of glycine, serine and methionine, epigenetic maintenance as well as providing antioxidant defense via glutathione (GSH) against RONS. Additionally, FOCM is also important in the generation of energy via adenosine triphosphate (ATP) generation in the mitochondria (Figure 1 and Figure 2).
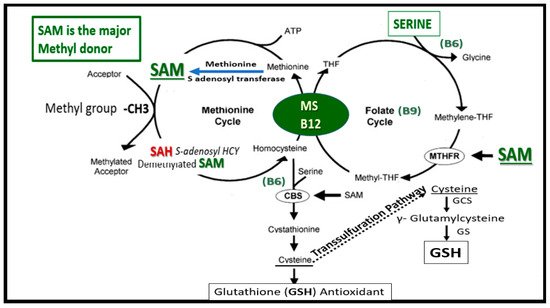
Figure 1. Folate-Mediated One-Carbon Metabolism (FOCM). This figure illustrates both the folate and methionine interdependent cycles and supports the importance of the methyl donor S-adenosylmethionine (SAM) as well as demonstrating the importance of the essential B vitamins. Importantly, note that methionine and tetrahydrofolate (THF) are derived primarily through dietary intake to supply the methionine and folate cycles and that the enzyme methionine synthase (MS) and its essential cofactor vitamin B12 are placed in a central position of the interconnected folate and methionine cycles. FOCM comprises a network of interconnected folate-dependent metabolic pathways responsible for serine and glycine interconversion, de novo purine synthesis, de novo thymidylate synthesis and homocysteine remethylation to methionine as well as providing antioxidant defense via glutathione (GSH) production via the transsulfuration pathway. Note that the encircled methylenetetrahydrofolate reductase (MTHFR) enzyme plays and important role in the folate cycle. The most common genetic variant in MTHFR gene to date is the 677C > T polymorphism, which results in elevated levels of Hcy especially if there is deficient folate. Once Hcy is synthesized through multiple steps in the methionine cycle, it may then undergo remethylation to methionine or be eliminated through the transsulfuration pathway. Additionally, thymidylate synthase (TYMS) converts deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) (not shown) in a 5,10-methylene-THF-dependent reaction. Importantly, cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE-CGL) do not only contribute to generate GSH (antioxidant) in the transsulfuration pathway but also are important for endothelial cell generation of hydrogen sulfide (H2S), a known gaso-transmitter and vasodilator. Elevation of Hcy from the methionine cycle may result in hyperhomocysteinemia, which is an independent risk factor for cerebro-cardiovascular diseases, accelerated atherosclerosis, thromboembolism, hypoxemia and stroke. CBS = cystathionine-beta-synthase; GCS = glutamate cysteine ligase (gamma-glutamylcysteine synthetase); GS = glutathione synthase; GSH = glutathione; MTHFR = methylenetetrahydrofolate; MS = methionine synthase; THF = tetrahydrofolate.
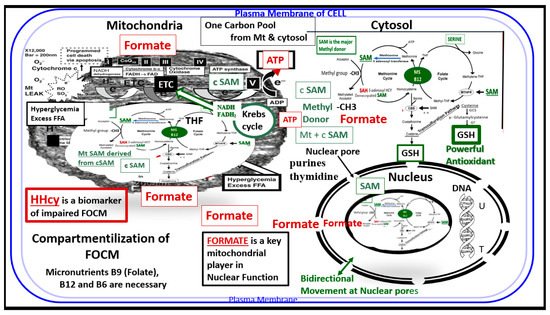
Figure 2. Compartmentalization of FOCM. Note the presence of the folate-methionine one carbon cycle metabolism in the cytoplasm (cytosol), mitochondria and nucleus. Additionally, note the importance of formate being transferred from the mitochondria to the nucleus, as well as S-adenosylmethionine (SAM) via nuclear pores. Importantly, deoxythymidine monophosphate (dTMP) synthesis occurs in the cytosol, nucleus and mitochondria, whereas purine synthesis and methionine synthesis take place within the cytosol. Mitochondrial FOCM generates formate for cytosolic and nuclear FOCM and biosynthetic precursors for mtDNA synthesis and mitochondrial protein translation. Thymidylate synthase (TYMS) converts deoxyuridine monophosphate (dUMP) to dTMP in a 5,10-methylene-THF-dependent reaction (not shown). It is important to note that mitochondrial SAM (Mt SAM) is derived from cytosolic SAM (cSAM). Additionally, the Krebs cycle also resides within the mitochondria and provides NADH and FADH2 to the electron transport chain for ATP production. ATP = adenosine triphosphate; c = cytosol; ETC = electron transport chain; FAD = flavin adenine dinucleotide; FADH = reduced flavin adenine dinucleotide; FFA = free fatty acids; HHcy = hyperhomocysteinemia; MS = methionine synthase; Mt = mitochondria; NADH = reduced nicotinamide adenine dinucleotide; T = thymidylate-thymine; U = uracil.
Additionally, the folate and methionine cycles are compartmentalized in cells and exist in the cytoplasm (cytosol), mitochondria and nucleus [12] (Figure 2)
Folate is necessary for FOCM, which involves an essential network of pathways involved in the transfer and eventual utilization of one-carbon units. This network is necessarily required for both DNA and RNA biosynthesis, amino acid metabolism, and methylation reactions. Deficient folate is most often related to the deficiency of the vitamins B9, B12 and B6. Note that when tetrahydrofolate (THF) enters the folate one-carbon cycle it acquires a carbon unit from serine (vitamin B6–dependent) to form 5,10 methyleneTHF, and once this is generated it follows along the folate cycle to result in various fates as in Figure 1 and Figure 2. Folate may also serve for conversion to 5-methyltetrahydrofolate (5-MTHF), or be utilized as the one-carbon donor in the synthesis of nucleic acids, where it is required by thymidylate synthetase in the conversion of deoyxuridine to deoxythymidine for pyrimidine biosynthesis or it may be converted to other metabolites for folate dependent purine biosynthesis [13][14]. While the focus in this review is primarily on B9, B12, and B6 there are also eight known B vitamins consisting of B1 thiamine, B2 riboflavin, B3 nicotinamide, B5 pantothenic acid, B6 pyridoxine, B7 biotin, B9 folate and B12 cobalamin that are important and necessary for proper homeostasis and health.
2. Impaired FOCM in T2DM
T2DM is a multifactorial polygenic disease that may be characterized as a chronic metabolic–endocrine disorder that associates with insulin resistance or relative lack of insulin or insulin deficiency and thus hyperglycemia. The resulting glucotoxic state promotes RONS stress and chronic inflammation [15].
Importantly, T2DM is an independent risk factor for macrovascular (accelerated atherosclerosis and vascular stiffness) and microvascular complications, which includes multiple diabetic-opathies such as retinopathy [16], neuropathy [17], nephropathy [16][17][18], vasculopathy—intimopathy [19], isletopathy [20], atheroscleropathy [21] and diabetic cognopathy (cognitive dysfunction) [22], which is associated with an increased risk for age-related neurodegenerative diseases such late onset Alzheimer’s disease (LOAD) [23].
T2DM is also associated with elevations of Hcy (HHcy) and thus, is associated with increased oxidative–redox stress as well as impaired FOCM [2][24][25][26][27]. Indeed, in the insulin resistant obese diabetic female db/db models there appears to be a strong and recurrent association of aberrant mitochondria (Mt) with enlargement and a loss of Mt matrix electron density with increased lucency and loss of crista that is associated with chromatin condensation in activated microglia and oligodenrocytes (Figure 3) [23][28][29][30].
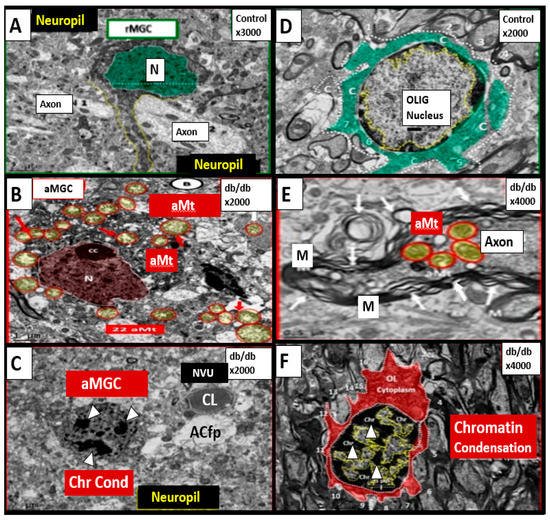
Figure 3. Chromatin Condensation in Aberrant Microglia and Oligodendrocytes in Diabetic Female db/db Models in Grey Matter—Cortical Layer III. This multipanel collage illustrates chromatin condensation in aberrant activated microglia cell(s) (aMGC) and aberrant oligodendrocytes (OL-OLG). Panels (A,D) illustrate the normal microglia cell (MGC) and oligodendrocyte in control non-diabetic models respectively. Note the abnormal aberrant mitochondria (aMt) in the microglia and neural cells in panels (B,E) respectively with aMt (highlighted yellow circles encircled in red). Panels (C,F) depict chromatin condensation (Chr Cond) within the nuclei of aberrant microglial cells and oligodendrocytes respectively. Also, note arrows in panel E depict myelin splitting and separation and arrowheads depict chromatin condensation in the nuclei (panels (C,F)). aMGCs and aberrant oligodendrocytes suggest abnormal crosstalk with abnormal myelin remodeling and impaired folate one-carbon metabolism. Magnification and scale bar varies. Images in this figure were provided and approved by CC 4.0 [28][29]. M = myelin; N = nucleus; NVU = neurovascular unit; Ol-Olig = oligodendrocyte.
Importantly, formate is primarily produced in the mitochondria and then secreted to enter the nucleus via nuclear pores in health to provide FOCM to the nucleus for proper chromatin, histone modeling and normal function (Figure 4) [31].
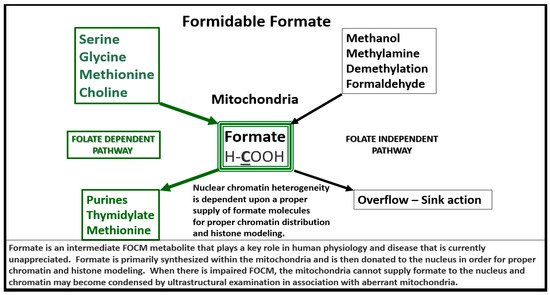
Figure 4. Formate Plays a Central and Formidable Role in Providing Proper Nucleus Function and Structure. Serine, glycine, methionine and choline are also necessary components to fulfill proper mitochondrial function to the nucleus in order to produce purines, thymidylate and methionine to fulfil their role in the nucleus. This figure depicts the importance of the folate dependent pathway (green coloring) for formate synthesis in the mitochondria and the central role for the formidable formate to be utilized by the nucleus for proper chromatin modeling. Note the bold and underlining of the important carbon unit in the chemical formula for formate. Also, note the folate independent pathway on the right-hand side of this figure. The concept for this slide design was derived from reference [31].
Therefore, aberrantly remodeled mitochondria as in the diabetic db/db models will not be able to produce formate and chromatin condensation ensues as noted in Figure 3. Importantly, Santos et al. recently demonstrated that deficiency of glycine (one of the precursors to formate in Figure 4) resulted in hydrocephalus—ventriculomegaly in glycine decarboxylase-deficient mice [32]. This is another finding suggesting the important role of FOCM and deficient formate involving abnormal brain structure. This group also suggested that impaired enzymatic activity within mitochondrial FOCM, as opposed to diminished exogenous supply or a methyl trap, can be a direct cause of aqueduct stenosis (the most commonly known cause of congenital hydrocephalus in humans).
The above findings regarding chromatin condensation in a diabetic model suggest an important role for FOCM in health and that impairment of FOCM is detrimental to physiologic homeostasis and normal development. Thus, impaired FOCM may play an important role in T2DM as more research is fostered in this exciting field of study. Additionally, Zhao et al. have recently shown that chronic folate deficiency induces glucose and lipid metabolism disorders and subsequent cognitive dysfunction in mice [33]. These authors also pointed out that previous studies have shown lower folate levels and higher Hcy levels in patients with T2D as compared with non-diabetic subjects [34][35]. Therefore, the involvement of impaired FOCM in T2DM seems to be viewed through the lens of HHcy, increased oxidative—redox stress and abnormal remodeling of mitochondria and nuclear chromatin condensation.
2.1. The Neurovascular Unit (NVU) in Diabetic Brain of the db/db Mouse
In our electron microscopy core laboratory, we have previously utilized the hyperleptinemic obese, insulin resistant and diabetic female db/db mouse models of diabetes in order to study the remodeling effects of diabetes in the brain [23][28][29][30]. These experiments in the db/db model have revealed a marked remodeling of the NVU. The cellular content of the control non-diabetic NVU includes the following four cells: the endothelial (EC), pericyte (Pc) and its foot processes (Pcfp), astrocyte (AC) and its foot processes (ACfp) and the microglia cell(s) (MGC) (Figure 5).
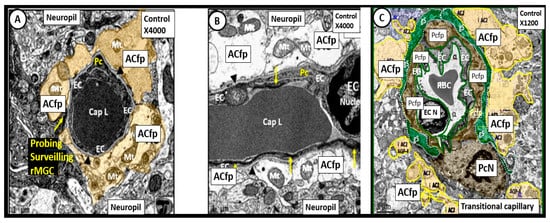
Figure 5. Neurovascular Unit Capillary (NVU) in control non-diabetic models. Panels (A) (cross section), (B) (longitudinal section) and (C) (cross section) depict the normal NVU composed of the endothelial cell (EC), pericyte foot processes (Pcfp), astrocyte foot processes (ACfp) and the ramified microglia cell (rMGC). Note in panel (A) that the rMGC is probing the NVU to surveil for any injury to the NVU. Note how tightly the Pcfp and the ACfp adhere and abut to the basement membrane of the ECs. Additionally, note the ACfp are pseudo colored yellow in panels (A,C) and reveal their electron lucent cytoplasm in panel (B), and the nanometer glymphatic space (gS) is pseudo colored green in panel (C). Varying magnifications upper right of each panel. Images are reproduced and modified by CC 4.0 [36].
In contrast to the control models the db/db diabetic models may be characterized by an attenuation and/or loss of the tight and adherens junctions (TJ/AJ) of the endothelial cells of the NVU, which form the electron dense barrier of the blood brain barrier (BBB). Importantly, we have been able to demonstrate an attenuation and or loss of the endothelial glycocalyx in the hypoleptinemic diabetic leptin deficient BTBR ob/ob models [36]. We also have observed Pcfp retraction and ACfp separation and retraction from the ECs of the NVU with microbleeds. Additionally, basement membrane thickening was also noted. Interestingly, we have observed that the ramified microglial cells (rMGCs) remodel to an ameboid—activated and invasive MGC that encompasses the NVU. These remodeling changes to the NVU allow for a leaky capillary endothelial NVU with increased permeability of the BBB as well as promoting NVU uncoupling in the cortex (layer III of the grey matter and hippocampus) (Figure 6) [36][37].

Figure 6. Compilation of Abnormal Remodeling in the Cortical Grey Matter Layer III in Diabetic db/db and BTBR ob/ob Neurovascular Unit (NVU). Panel (A) is the control model and depicts the normal morphology of the NVU and the remainder of images are compiled from the diabetic db/db and BTBR ob/ob models. Panel (B1) (control with intact blood-brain barrier (BBB)) and (2–4) depicts the attenuation and or loss of the endothelial cell (EC) tight and adherens junctions (TJ/AJ) of the BBB. Panel (C1) depicts the highly electron dense endothelial glycocalyx (arrows) found in non-diabetic control models while Panel (2) depicts the attenuation and/or loss of the endothelial glycocalyx (arrows) by lanthanum nitrate perfusion fixation staining in the BTBR ob/ob diabetic model. Panel (D) depicts astrocyte foot process detachments from the endothelial neurovascular unit (NVU) with a nearby activated microglial cell (aMGC) and a labeled microbleed adjacent to the NVU in this low magnification image. Panel (E) depicts marked basement membrane thickening that is associated with the capillary NVU and note the detached astrocyte foot processes (ACfp) pseudo-colored red and the aMGC. Panel (F) depicts an aMGC (pseudo-colored red) that is encompassing the capillary NUV and note again the detached and retracted ACfp (drAC) (pseudo-colored cyan) with magnification ×2500 (not shown). These images display the remodeling changes that accompany the uncoupling of the NVU in brain grey matter in cortical layers III. Images in this compilation figure were modified with permission by CC 4.0 [23][28].
The uncoupling of the NVU results in regional hypoxia and decreased cerebral blood flow (CBF) with associated impaired neurotransmission and impaired cognition. Other than the chromatin condensation in activated microglia, we do not presently know if impaired FOCM is directly involved in NVU remodeling and uncoupling; however, attenuation and/or loss of the EC TJ/AJ due to increased RONS may be due to impaired FOCM and a decrease in the antioxidant glutathione (GSH) as a result of HHcy. This increase in RONS could definitely contribute to the changes in the morphological remodeling of the NVU cells and could also increase the permeability of the NVU and its neurovascular uncoupling due to elevated levels of Hcy—HHcy (in impaired FOCM) in addition to the elevations in glucose (glucotoxicity) and oxidative—redox stress with increased RONS due to T2DM. Importantly, glucotoxicity and HHcy may act synergistically due to the increased RONS due to both glucotoxicity induced RONS and HHcy induced RONS.
2.2. The Endothelial Glycocalyx (ecGCx) in T2DM
The intact endothelial glycocalyx (ecGCx) is important for the vascular integrity of arteries, arterioles and capillaries. It is composed of a sugar-protein mesh, gel-like surface coating on the luminal apical polarized endothelial monolayer. This protective surface layer coating modulates the direct contact of the blood and its components (circulating leukocytes, platelets, red blood cells and larger plasma proteins) with the extracellular matrix ecGCx surface layer of the endothelial cells. The ecGCx is primarily synthesized by the endothelium with some contributions by plasma albumin, orosomucoids, fibrinogen, circulating glycoproteins and glycolipids [23][36][37][38][39][40][41][42][43][44]. Additionally, the ecGCx is anchored to the endothelial luminal plasma membranes by highly sulfated proteoglycans (syndecans and glycipans), glycoproteins (including selectins such as various cellular adhesion molecules and integrins) along with non-sulfated hyaluronan (a glycosaminoglycan) via CD44. Hyaluronan may also be free floating (unbound) or attached to the assembly proteins, such as the endothelial hyaluronan synthases as well as forming hyaluronan-hyaluronan unbound stable complexes.
Importantly, mechanotransduction is accomplished by the ecGCx via the anchored proteoglycan (glypican) to the caveolae [45][46]. The ecGCx is important for both the overall barrier function of the endothelium and to the mechanotransduction of endothelial fluid shear stress. Luminal shear stress induces the production of endothelial derived nitric oxide (NO) via lipid rafts of the caveolae and the glycoproteins of the ecGCx to produce shear stress-induced nitric oxide (NO). NO production is very important for vasodilation of the aorta and arterioles via the signaling of NO to the vascular smooth muscle cells and pericytes of the capillaries. If this precious mechanism is lost due to a disturbance, attenuation, loss or shedding of the ecGCx then there will be a decrease in the quintessential bioavailable NO to signal vascular smooth muscle cells in arteries, arterioles and pericyte capillaries. An attenuation or loss of bioavailable NO promotes regional ischemia for tissue beds in all organs (Figure 7).
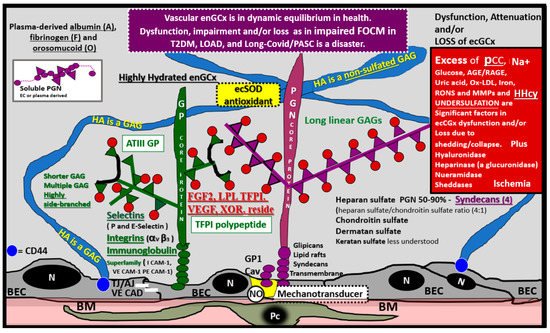
Figure 7. The Endothelial Glycocalyx (ecGCx). This illustration depicts the normal components of the ecGCx: a unique extracellular matrix. The ecGCx in T2DM, LOAD and Long COVID/PASC is the first component of the endothelial cell (EC) that comes into contact with the blood components of the vascular lumen. The normal components of the ecGCx include two classes of proteins that are mostly anchored proteoglycans (PGN) (purple), glycoproteins (GP) (green) and Hyaluronic acid—Hyaluronan (HA) (blue) that is an exceedingly long polymer of disaccharides that are non-sulfated glycosaminoglycans. HA may be either unattached—free floating or anchored to CD44 on the plasma membrane of ECs, or form HA–HA complexes. HA may also reversibly interact at the lumen with plasma-derived albumin, fibrinogen and soluble PGNs. The PGNs and GPs side chains consist of glycosaminoglycans (GAGs), which are covalently bound to their core proteins and are highly sulfated, which are important for giving the ecGCx its net-negative charge. The two primary PGNs are the syndecans and glypicans. The GPs consist primarily of selectins (P and E), integrins (alpha v and beta 3) and the immunoglobulin superfamily of ICAM-1, VE-CAM and PE CAM-1. Caveolae are invaginations of lipid rafts on the EC plasma membrane and contain CD44 important to anchor glycosylphosphatidylinositol (GPI) that anchor glypican-1. The GPI/glypican-1 interaction is thought to activate endothelial nitric oxide synthase (eNOS) to produce bioavailable nitric oxide (NO) via the calcium calmodulin dependent Caveolin-1 (Cav-1) protein. Note on the right-hand side of this image the numerous causes for the attenuation/shedding or loss of the ecGCx. Note that Hcy is included since it is elevated in both T2DM and Late onset Alzheimer’s disease (LOAD). Hcy may compromise the ecGCx due to its elevation, which results in hyperhomocysteinemia (HHcy), oxidative stress with elevation in reactive oxygen nitrogen species (RONS), inflammation and activation of matrix metalloproteinases. Note that T2DM is known to increase hyaluronidase. The impaired FOCM with hyperhomocyteinemia, oxidative stress and inflammation can be damaging to the ecGCx and contribute to endothelial cell activation and dysfunction with detrimental effects on the vascular tissue that predispose to increased vascular inflammation and a prothrombotic state and ischemia, which is also an inducer of ecGCx loss. Image provided by CC 4.0 [44]. A = albumin; AGE/RAGE = advanced glycation end products and receptor to AGE; N = nucleus; ATPIII GP = antithrombin three glycoprotein; BEC = brain endothelial cell or just endothelial cell; BM = basement membrane; CAD = cadherin; CAM = cellular adhesion molecule; CD44 = cluster of differentiation 44; ecSOD = extracellular superoxide dismutase; F = fibrinogen; FGF2 = fibroblast Growth Factor 2; FOCM = folate-mediated one-carbon metabolism; GCx = glycocalyx; ICAM-1 = intercellular adhesion molecule; Ox LDL = oxidized low-density lipoprotein; LPL = lipoprotein lipase; MMPs = matrix metalloproteinases; N = nucleus; Na+ = sodium; O = orosomucoids; Pc = vascular mural cell pericyte(s); PECAM-1 = platelet endothelial cell adhesion molecule-1. RONS = reactive oxygen species; TFPI = tissue factor pathway inhibitor; TJ/AJ = tight and adherens junctions; VCAM = Vascular cell adhesion protein; VE CAD = vascular endothelial cadherins; VEGF = vascular endothelial growth factor; XOR = xanthine oxioreductase.
Recently, duPreez et al. has shared that the degree of sulfation and/or the position of sulfate groups may result in undersulfation and dysfunction of glycosaminoglycans of the proteoglycans and glycoproteins of the glycocalyx [47]. This group has also suggested that undersulfation and the improper positioning of sulfate groups may increase the susceptibility to COVID-19 and its severity, and therefore possibly LC/PASC, since we now know that the more severe cases of the acute disease predispose to increases in LC/PASC. Thus, the undersulfated glycocalyx may not only increase susceptibility to SARS-CoV-2 infections but also could result in increased inflammation, vascular permeability with shedding of the ecGCx that could additionally give rise to procoagulant and antifibrinolytic states and possible multiple organ failure as occurs in SARS-CoV-2 [47]. Glycocalyx degradation, attenuation, shedding and or loss is gaining recognition as an important aspect in multiple diseases, including T2DM, LOAD and all forms of sepsis including COVID-19 pathophysiology and possibly even LC/PASC.
2.3. Aberrant Mitochondria and Impaired FOCM in T2DM
The mitochondria play an important role not only in energy production but also an important role in FOCM. In Section 1 (Figure 2) the compartmentalization of FOCM was presented showing that the mitochondrial and cytosolic FOCM operate in a parallel fashion to contribute to the formation of formate by the mitochondria and to donate SAM and tetrahydrofolate (THF) to the nucleus. Thus, the importance of the mitochondria FOCM must remain intact. If there is aberrant morphological remodeling of the mitochondria (aMt), it cannot (along with the parallel functions of the cytosol) deliver the proper molecules (formate, SAM and THF) to the nucleus in order to carry out its role in maintaining nuclear DNA and RNA modeling to allow for normal cellular function. In the female diabetic db/db models, we have observed marked abnormal remodeling of the mitochondria in each of the brain cells (Figure 8).
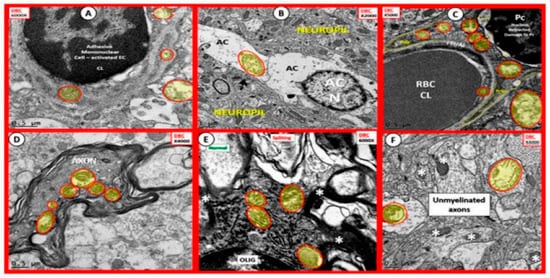
Figure 8. Aberrant Mitochondria in Brain Endothelial cells (EC), Astrocytes (AC), Pericytes (Pc), Myelinated and Unmyelinated Neurons and Oligodendrocytes (OLIG) in the Diabetic db/db Models. The aberrant mitochondria (aMt) are pseudo-colored yellow with red outlines in order to allow for rapid identification. Panels (A–F) demonstrate aberrant mitochondria (aMt) in each of the brain cells depicted. Panel (A) depicts aMt in ECs and adjacent AC foot processes. Panel (B) depicts an aMt within the cytoplasm of an AC. Panel (C) depicts multiple aMt in the cytoplasm of a Pc. Panel (D) depicts aMt within a myelinated neuronal axon that also demonstrates separation of its lining myelin sheath. Panel (E) depicts aMt in an oligodendrocyte’s cytoplasm. Panel (F) depicts aMt with the axoplasm of an unmyelinated neuronal axon. Note that most of the aMt are hypolucent as a result of the loss of their normal electron dense matrix proteins and they share a common feature of attenuated and or loss of their crista; they are also enlarged. Magnifications are located in the upper part of each panel and the scale bars are at the bottom. The images in this figure were modified with permission by CC 4.0 [23].
As noted in Figure 8, the markedly remodeled aMt in the diabetic db/db models would be incapable of proper mitochondrial FOCM function and could not function in parallel with the cytosolic FOCM in order to donate formate, SAM and THF to the nucleus to properly function. Importantly, these aMt remodeling changes could also place the nucleus at risk for epigenetic modifications and/or mutations, which could impair proper nuclear function. Additionally, T2DM is known to have elevated Hcy in humans and in the db/db diabetic models and this elevation of Hcy is thought to be a biomarker of impaired FOCM. Currently, it is not known which comes first: the abnormal structural remodeling to the mitochondria due to excess energy (hyperglycemia and elevated free fatty acid due to T2DM that remodel the Mt or if this is combined with the impaired mitochondria FOCM that could result in the Mt DNA abnormalities and loss of Mt matrix proteins as noted on the transmission electron micrographs in Figure 8; it may be that it is a bit of both working in concert to result in abnormal structure and function.
This entry is adapted from the peer-reviewed paper 10.3390/medicina58010016
References
- Mason, J.B. Biomarkers of Nutrient Exposure and Status in One-Carbon (Methyl) Metabolism. J. Nutr. 2003, 133 (Suppl. 3), 941S–947S.
- Hayden, M.R.; Tyagi, S.C. Homocysteine and reactive oxygen species in metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: The pleiotropic effects of folate supplementation. Nutr. J. 2004, 3, 4.
- Crapser, J.D.; Spangenberg, E.E.; Barahona, R.A.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N. Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 2020, 58, 102919.
- Barroso, M.; Handy, D.E.; Castro, R. The Link between Hyperhomocysteinemia and Hypomethylation: Implications for Cardiovascular Disease. J. Inborn Errors Metab. Screen. 2017, 5, 1–15.
- Herrmann, W. Significance of hyperhomocysteinemia. Clin. Lab. 2006, 52, 367–374.
- Graham, I.M.; O’Callaghan, P. Vitamins, homocysteine and cardiovascular risk. Cardiovasc. Drugs Ther. 2002, 16, 383–389.
- Ponti, G.; Ruini, C.; Tomasi, A. Homocysteine as a potential predictor of cardiovascular risk in patients with COVID-19. Med. Hypotheses 2020, 143, 109859.
- Ibrahimagić, O.Ć.; Smajlović, D.; Dostović, Z.; Vidović, M.; Tupković, E.; Kunić, S. COMMENT ON AN ARTICLE: “Homocysteine as a potential predictor of cardiovascular risk in patients with COVID-19”. Med. Hypotheses 2020, 143, 110107.
- Boers, G.H. Mild hyperhomocysteinemia is an independent risk factor of arterial vascular disease. Semin. Thromb. Hemost. 2000, 26, 291–295.
- Blom, H.J. Consequences of homocysteine export and oxidation in the vascular system. Semin. Thromb. Hemost. 2000, 26, 227–232.
- Jakubowski, H.; Zhang, L.; Bardeguez, A.; Aviv, A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: Implications for atherosclerosis. Circ. Res. 2000, 87, 45–51.
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42.
- Bailey, L.B.; Stover, P.J.; McNulty, H.; Fenech, M.F.; Gregory, J.F.; Mills, J.L.; Pfeiffer, C.M.; Fazili, Z.; Zhang, M.; Ueland, P.M.; et al. Biomarkers of nutrition for development- folate review. J. Nutr. 2015, 1447, 1636S–16380S.
- McNulty, H.; Strain, J.J.; Hughes, C.F.; Pentieva, K.; Ward, M. Evidence of a Role for One-Carbon Metabolism in Blood Pressure: Can B Vitamin Intervention Address the Genetic Risk of Hypertension Owing to a Common Folate Polymorphism? Curr. Dev. Nutr. 2019, 4, nzz102.
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63.
- Yang, Y.; Hayden, M.R.; Sowers, S.; Bagree, S.V.; Sowers, J.R. Retinal redox stress and remodeling in cardiometabolic syndrome and diabetes. Oxid. Med. Cell Longev. 2010, 3, 392–403.
- Hayden, M.R.; Salam, M.; Sowers, J.R. Reactive Oxygen Species and Diabetic Peripheral Neuropathy—A Closer Look, Chapter 149; Lahey, I., Ed.; Systems Biology of Reactive Oxygen Species and Antioxidants; Springer: Berlin/Heidelberg, Germany, 2014; pp. 3375–3400.
- Hayden, M.R.; Whaley-Connell, A.; Sowers, J.R. Renal redox stress and remodeling in metabolic syndrome, type 2 diabetes mellitus, and diabetic nephropathy: Paying homage to the podocyte. Am. J. Nephrol. 2005, 25, 553–569.
- Hayden, M.R.; Tyagi, S.C. Intimal redox stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Cardiovasc. Diabetol. 2002, 1, 3.
- Hayden, M.R.; Sowers, J.R. Isletopathy in Type 2 diabetes mellitus: Implications of islet RAS, islet fibrosis, islet amyloid, remodeling, and oxidative stress. Antiox Redox Signal. 2007, 9, 891–910.
- Hayden, M.R.; Tyagi, S.C. Is type 2 diabetes mellitus a vascular disease (atheroscleropathy) with hyperglycemia a late manifestation? The role of NOS, NO, and redox stress. Cardiovasc. Diabetol. 2003, 2, 2.
- Hayden, M.R.; Banks, W.A.; Shah, G.N.; Gu, Z.; Sowers, J.R. Cardiorenal metabolic syndrome and diabetic cognopathy. Cardiorenal. Med. 2013, 3, 265–282.
- Hayden, M.R. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262.
- Joshi, M.B.; Baipadithaya, G.; Balakrishnan, A.; Hegde, M.; Vohra, M.; Ahamed, R.; Nagri, S.K.; Ramachandra, L.; Satyamoorthy, K. Elevated homocysteine levels in type 2 diabetes induce constitutive neutrophil extracellular traps. Sci. Rep. 2016, 6, 36362.
- Huang, T.; Ren, J.; Haung, J.; Li, D. Association of homocysteine with type 2 diabetes: A meta-analysis implementing Mendelian randomization approach. BMC Genom. 2013, 14, 867.
- Finer, S.; Saravanan, P.; Hitman, G.; Yajnik, C. The role of the one-carbon cycle in the developmental origins of Type 2 diabetes and obesity. Diabet. Med. 2014, 31, 263–272.
- Mursleen, M.T.; Riaz, S. Implication of homocysteine in diabetes and impact of folate and vitamin B12 in diabetic population. Diabetes Metab. Syndr. 2017, 11 (Suppl. 1), S141–S146.
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part II: Microglia and Mitochondria. Neuroglia. Neuroglia 2018, 1, 311–326.
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model–Part III: Oligodendrocyte and Myelin. Neuroglia 2018, 1, 351–364.
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Empagliflozin Ameliorates Type 2 Diabetes-Induced Ultrastructural Remodeling of the Neurovascular Unit and Neuroglia in the Female db/db Mouse. Brain Sci. 2019, 9, 57.
- Pietzke, M.; Meiser, J.; Vazquez, A. Formate metabolism in health and disease. Mol. Metab. 2020, 33, 23–37.
- Santos, C.; Pai, Y.J.; Mahmood, M.R.; Leung, K.Y.; Savery, D.; Waddington, S.N.; Copp, A.J.; Greene, N. Impaired folate 1-carbon metabolism causes formate-preventable hydrocephalus in glycine decarboxylase-deficient mice. J. Clin. Investig. 2020, 130, 1446–1452.
- Zhao, M.; Yuan, M.M.; Yuan, L.; Huang, L.L.; Liao, J.H.; Yu, X.L.; Su, C.; Chen, Y.H.; Yang, Y.Y.; Yu, H.; et al. Chronic folate deficiency induces glucose and lipid metabolism disorders and subsequent cognitive dysfunction in mice. PLoS ONE 2018, 13, e0202910.
- Wang, D.; Zhai, J.X.; Liu, D.W. Serum folate, vitamin B12 levels and diabetic peripheral neuropathy in type 2 diabetes: A meta-analysis. Mol. Cell Endocrinol. 2017, 443, 72–79.
- Wee, A.K. Serum folate predicts muscle strength: A pilot cross-sectional study of the association between serum vitamin levels and muscle strength and gait measures in patients >65 years old with diabetes mellitus in a primary care setting. Nutr. J. 2016, 15, 89.
- Hayden, M.R.; Banks, W.A. Deficient Leptin Cellular Signaling Plays a Key Role in Brain Ultrastructural Remodeling in Obesity and Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 5427.
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part I: Astrocyte. Neuroglia 2018, 1, 220–244.
- Luft, J.H. Fine structures of capillary and endocapillary layer as revealed by ruthenium red. Fed. Proc. 1966, 25, 1773–1783.
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. 2007, 454, 345–359.
- Haeren, R.H.L.; Rijkers, K.; Schijns, O.E.M.G.; Dings, J.; Hoogland, G.; van Zandvoort, M.A.M.J.; Vink, H.; van Overbeeke, J.J. In vivo assessment of the human cerebral microcirculation and its glycocalyx: A technical report. J. Neurosci. Methods 2018, 303, 114–125.
- Yoon, J.H.; Jeong, Y. In vivo imaging for neurovascular disease research. Arch. Pharm. Res. 2019, 42, 263–273.
- Reed, M.J.; Damodarasamy, M.; Banks, W.A. The extracellular matrix of the blood-brain barrier: Structural and functional roles in health, aging, and Alzheimer’s disease. Tissue Barriers 2019, 7, 1651157.
- Hayden, M.R. Endothelial activation and dysfunction in metabolic syndrome, type 2 diabetes and coronavirus disease 2019. J. Int. Med. Res. 2020, 48, 300060520939746.
- Hayden, M.R. Hypothesis: Neuroglia Activation Due to Increased Peripheral and CNS Proinflammatory Cytokines/Chemokines with Neuroinflammation May Result in Long COVID. Neuroglia 2021, 2, 7–35.
- Pahakis, M.Y.; Kosky, J.R.; Dull, R.O.; Tarbell, J.M. The role of endothelial glycocalyx components in mechanotransduction of fluid shear stress. Biochem. Biophys. Res. Commun. 2007, 355, 228–233.
- Tarbell, J.M.; Pahakis, M.Y. Mechanotransduction and the glycocalyx. J. Int. Med. 2006, 259, 339–350.
- du Preez, H.N.; Aldous, C.; Hayden, M.R.; Kruger, H.G.; Lin, J. Pathogenesis of COVID-19 described through the lens of an undersulfated and degraded epithelial and endothelial glycocalyx. FASEB J. 2022, 36, e22052.
This entry is offline, you can click here to edit this entry!