Chronic hepatitis B virus (HBV) infection is one of the most common factors associated with hepatocellular carcinoma (HCC), which is the sixth most prevalent cancer among all cancers worldwide. The mechanisms underlying HBV-induced malignant transformation remain unclear, but some studies have suggested that the hepatitis B virus core (HBc) protein has a potential function in the pathogenesis of hepatocellular carcinoma in addition to the HBV X protein. This review focuses its discussion on the involvement of HBc in the development of hepatocellular carcinoma.
- hepatitis B virus
- hepatitis B virus core protein
- hepatocellular carcinoma
- hepatocarcinogenesis
1. Introduction
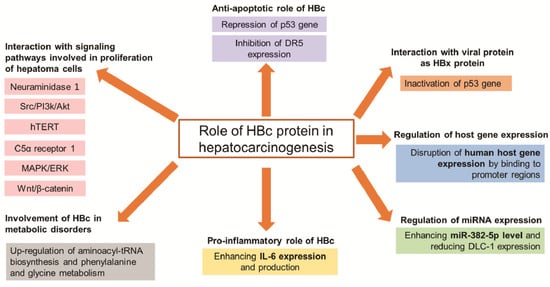
| Group | Target | Mechanism of HBc to Promote Hepatocarcinogenesis |
Reference |
|---|---|---|---|
| Signaling pathways | Neuraminidase 1 | Promote NEU1 expression inducing proliferation and migration of hepatoma cells | [18] |
| Src/PI3k/Akt | Activate Src/PI3k/Akt pathway inducing tumor formation of hepatoma cells | [14] | |
| hTERT | Upregulate the c-Ets2-dependent expression of hTERT inducing hepatoma cell proliferation | [17] | |
| C5α receptor 1 | Upregulate C5AR1 via NF-κB pathway to facilitate the growth and migration of hepatoma cells | [19] | |
| MAPK/ERK and Wnt/β-catenin | Bind to gene promoters of these pathways, thus participating in the progression of HCC | [20] | |
| Anti-apoptosis | p53 | Prevent hepatoma cells from anti-Fas antibody-induced apoptosis through the p53-dependent Fas/FasL signaling pathway | [21] |
| Repress the p53 gene through the transcription factor E2F1 binding site in the p53 promoter | [22] | ||
| DR5 | Prevent hepatocytes from TRAIL-induced apoptosis through inhibiting DR5 expression | [23] | |
| Metabolic disorders | Cell metabolism | Upregulate aminoacyl-tRNA biosynthesis and phenylalanine and glycine metabolism inducing development of HCC | [24] |
| Immune system |
IL-6 | Enhance IL-6 expression and production that involved in pathogenesis of HBV | [25] |
| Epigenetic | miR-382-5p | Promote HCC metastasis through enhancing miR-382-5p level and reducing DLC-1 expression | [26] |
| Genetic | Promoter regions |
Disrupt human host gene expression by binding to promoter regions, which modulate normal functions of liver cells | [20] |
| Interaction with viral protein as HBx protein | p53 | Inactivate the p53 gene, thus participating in HCC progress | [22] |
NEU1: neuraminidase 1; hTERT: human telomerase reverse transcriptase; C5AR1: C5α receptor 1; MAPK/ERK: mitogen-activated protein kinase/extracellular signal-regulated kinase; DR5: death receptor 5; MKK7: mitogen-activated protein kinase kinase 7; IL-6: interleukine-6; HBV: hepatitis B virus; HCC: hepatocellular carcinoma; DLC-1: deleted in liver cancer.
2. HBc Protein, Description, and Functions in the Viral Life Cycle
3. HBc Protein, a Pleiotropic Role in Hepatocarcinogenesis
3.1. Interaction with Signaling Pathways Involved in Proliferation of Hepatoma Cells
3.1.1. Neuraminidase 1 Pathway
3.1.2. Abnormal Sarcoma (Src)/PI3k/Akt Pathway
3.1.3. Human Telomerase Reverse Transcriptase (hTERT) Pathway
3.1.4. C5α Receptor 1 Pathway
3.1.5. Mitogen-Activated Protein Kinase (MAPK)/ERK and Wnt/β-Catenin Pathways
3.2. HBc Protein, an Anti-Apoptotic Viral Protein
Resistance of HBV-infected hepatocytes to apoptosis is considered one of the major causes in the progression of chronic hepatitis to cirrhosis and ultimately to HCC [23][73]. Apoptosis of HBV-infected hepatocytes is mainly mediated by signaling belonging to the tumor necrosis factor (TNF) protein family, including TNF-α, Fas ligand (FasL), and TNF-related apoptosis-inducing ligand (TRAIL) [74][75]. TNF-α and FasL are considered as death receptor ligands.
3.2.1. Repression of the Proapoptotic p53
FasL induces apoptosis of hepatocytes in both normally functioning liver and in various forms of liver disease [76] and the Fas/FasL system plays an important role in hepatocyte death during HBV infection. In response to DNA damage, the p53 tumor suppressor protein induces either apoptosis or cell cycle arrest at the G1-S. Previous studies reported that Fas transcriptional expression is regulated by p53 protein in hepatoma cells, and the cross-talk between the p53 and Fas-FasL pathways in modulating apoptosis is clinically important [77][78]. HCC may progress through the deactivation of the p53 gene [79][80].
A study demonstrated that the expression of HBx protein in infected cells inhibits the induction of apoptosis by direct interaction with the tumor suppressor p53 [81]. A similar mechanism is observed with HBc protein. Liu et al. therefore found that HBc mediated resistance of human hepatoma cells to agonistic anti-Fas antibody-induced apoptosis. They then identified that HBc significantly downregulated the expression of p53, total Fas and membrane-bound Fas at the RNA and protein levels and reduced FasL at the transcriptional level. In contrast, HBc increased the expression of soluble forms of Fas (sFas) by facilitation of Fas alternative splicing. Mechanistically, HBc-mediated Fas alternative mRNA splicing was associated with the upregulation of polypyrimidine tract-binding protein 1 and the downregulation of Fas-activated serine/threonine kinase. HBc may prevent hepatocytes from apoptosis induced by Fas/FasL system by the dual effects of reducing the expression of the proapoptotic form of Fas and enhancing the expression of the antiapoptotic form of the receptor, which may contribute to the survival and persistence of infected hepatocytes toward the development of chronic HBV infection. HBc is a survival factor capable of protecting cells, such as hepatoma cells from anti-Fas antibody-induced apoptosis through the p53-dependent Fas/FasL signaling pathway [21].
The E2F family of transcription factors plays an essential role in mediating cell cycle progression, particularly those involved in G1-S progression [82], and it has been implicated in the regulation of growth inhibition, differentiation, apoptosis, and oncogenic transformation. Previous studies have shown that E2F1 functions as both an oncogene and a tumor suppressor gene [83][84]. A study revealed that HBc protein is a transcriptional repressor of the human p53 gene. Indeed, an electrophoretic mobility shift assay demonstrated that the binding of HBc to E2F1 reduced the DNA-binding ability of E2F1 at the p53 promoter [22].
3.2.2. TNF-Related Apoptosis-Inducing Ligand (TRAIL) Apoptotic Pathway
Unlike TNF and FasL, TRAIL preferentially induces apoptosis of tumor cells and virus-infected cells, but does not induce apoptosis of normal cells [75][85][86]. Following high-risk human papillomavirus (HPV) infection, viral proteins use different strategies to modulate apoptosis. In particular, the E5 protein of HPV can disrupt TRAIL-mediated apoptosis, which suggests that it may prevent apoptosis of cells at early stages of viral infection [87].
The TRAIL was recently reported to be implicated in hepatocyte death during HBV infection. Interestingly, two HBV proteins, HBx and truncated middle hepatitis B surface protein (MHBs(t)), were found to sensitize hepatocytes to TRAIL-induced apoptosis. Other data showed that HBx enhances TRAIL-induced apoptosis through Bax upregulation, whereas MHBs(t) does this through ERK2 activation [75][88].
In contrast, Liu et al. found that HBc protein had an opposite role in TRAIL-induced hepatocyte apoptosis. Indeed, HBc would be a strong inhibitor of TRAIL-induced apoptosis by blocking death receptor 5 (DR5) expression [89] inducing a decrease of TRAIL-induced apoptosis of human hepatoma cells. The DR5 gene promoter has no typical TATA-box, but has two Sp1 sites responsible for the basal transcription activity of the DR5 gene [90]. Transcription factors such as NF-kB and p53 can regulate the DR5 promoter activity. In hepatoma cell lines expressing the core protein, HBc protein induces a significant reduction in DR5 expression that represses the DR5 promoter activity. Consequently, HBc prevents hepatocytes from TRAIL-induced apoptosis through inhibiting DR5 expression [23]. In practice, if the pro-apoptotic proteins, such as HBx, are majority, HBV-infected hepatocytes may die as a consequence, and fulminant hepatitis may develop. In contrast, if the anti-apoptotic viral proteins, such as HBc predominate, the infected hepatocytes may not undergo apoptosis and chronic HBV infection may ensue.
3.3. HBc Protein, a Pro-Apoptotic Viral Protein
HBc protein would prevent hepatocyte from FasL-induced apoptosis by altering the membrane and soluble Fas level, while it would also prevent sensitized TNF-α-induced apoptosis by disrupting the interaction between mitogen-activated protein kinase kinase 7 (MKK7) and receptor of activated protein kinase C 1 (RACK1). RACK1 is described as a scaffold protein that facilitates the phosphorylation of MKK7 by its upstream activators.
One study with ectopic expression of HBc in HepG2 cells and primary hepatocyte cultures reported that HBc abolishes the interaction between MKK7 and RACK1 by competing with MKK7 for binding to RACK1, thereby downregulating TNF-induced phosphorylation of MKK7 and the activation of JNK, an important regulator of TNF-α signaling. Specific knockdown of MKK7 increases the sensitivity of hepatocytes to TNF-induced apoptosis, while overexpression of RACK1 counteracts the proapoptotic activity of HBc. The expression of HBc makes hepatocytes susceptible to TNF-induced apoptosis by disrupting the interaction between MKK7 and RACK1 [91] and this finding suggests a direct role of the core protein in driving liver pathogenesis in chronically infected patients.
3.4. HBc Protein, a Viral Protein Involved in Metabolic Disorders
The progression of cancer seems to involve major disorders in cell metabolism [92]. Metabolic disorders are shared by both transformed cells and those infected with viruses, suggesting that metabolic reprogramming is an important hallmark of viral oncogenesis. Viruses handle metabolic pathways and associated-signaling cascades to provide sufficient resources for the production of new virions. Among viruses, chronic Hepatitis C virus (HCV) infection is more associated with metabolic alterations than HBV infection. Indeed, patients with chronic HCV often develop secondary metabolic disorders, such as insulin resistance and steatosis [93].
One study reported that the link between metabolic disorders and HCC could be attributed to the effects of HBV infection and in particular the HBV-encoded proteins [94], such as HBx protein [95]. Recently, multi-omics analyses of HBc transfected cells revealed that HBc protein promotes the expression of multiple metabolic enzymes and the secretion of metabolites from hepatoma cells modifying the metabolic characteristics of HCC cells, and contributes to HBV-related metabolic dysregulation through the modulation of glycolysis and amino acid metabolism. For instance, glycolysis and amino acid metabolism are significantly upregulated by HBc. Max-like protein X (MLX) would be an important protein in glycolysis and lipid biosynthesis in tumorigenesis. Besides, MLX might be recruited and enriched by HBc in the nucleus to regulate glycolysis pathways. Moreover, PGK1 is also upregulated by HBc. A recent study highlighted that PGK1 acted as a protein kinase in coordinating glycolysis and the tricarboxylic acid cycle, which is instrumental in cancer metabolism and tumorigenesis [96]. Therefore, Xie et al. concluded that nine pathways were considered closely related to the development of HCC, including aminoacyl-tRNA biosynthesis and phenylalanine and glycine metabolism [24].
Dysregulated cholesterol homeostasis is a characteristic of numerous diseases, including liver fibrosis, and even many cancers. A recent study showed that ethanol and HBV together synergistically enhance cholesterol biosynthesis and decrease cholesterol utilization and its uptake in vivo and in vitro. Thus, HBV is involved in the dysregulation of cholesterol homeostasis and increases hepatic cholesterol deposition in alcoholic fatty liver via the hepatitis B core protein [97]. These changes may contribute to the progression of various coexisting diseases.
3.5. HBc Protein, a Pro-Inflammatory Viral Protein
Interleukin (IL)-6 is one of the most significant cytokines involved in hepatic inflammation and hepatocarcinogenesis in patients with liver diseases [98][99][100]. Its role has been described in HCV [101] and HBV infections [100]. Higher serum IL-6 level was an independent risk factor for HCC development in female hepatitis C patients. In the case of HBV infection, high serum IL-6 level was also associated with HCC risk [98] and aspartate aminotransferase [102], and considered as a prognostic indicator in HCC [103]. Previous studies described that human hepatoma cells and hepatic cells secrete IL-6 after activating NF-κB pathway and a MyD88-dependent signaling pathway, whose activation is regulated by protein phosphatase type 2 C alpha in the presence of HBx protein [104][105][106]. An intracellular HBcAg expression model (transfected hepatocyte-like cells) showed that the expression of HBc in hepatocytes enhances IL-6 expression and production (checked by qPCR and ELISA, respectively), which was mediated through activating p38 mitogen-activated protein kinase, extracellular signal-related kinase and NF-κB pathways. Cytoplasmic HBcAg seems to be a viral antigen for immune-mediated liver damage, and HBV-infected parenchymal cells may produce proinflammatory cytokines that are involved in pathogenesis of hepatitis B [25].
3.6. HBc Protein, a Regulator of miRNA Expression
Accumulated epigenetic alterations including histone modification, DNA methylation and non-coding RNA (micro RNA or miRNA, lncRNA) were described to have profound significance in HBV-related carcinogenesis [107]. Through its partially complementary sequence to the 3′-UTR of target mRNAs, miRNAs result in gene silencing via translational repression and/or mRNA degradation and are therefore involved in regulating almost all known physiological and pathological processes [108]. miRNAs play a key role in host-virus interactions [109] and their dysregulation is involved in liver fibrosis and a number of human cancers such as HCC. MiR-122 is the most abundant miRNA in the liver, representing 70% of the total miRNA in hepatocytes [110]. MiR-122 is downregulated in patients with HBV-related HCC, while it is upregulated in patients with HBV chronic infection [111].
HBc protein promotes the hepatocarcinogenesis process through the regulation of some miRNAs. The deleted in liver cancer (DLC-1) gene encodes a Rho-GTPase activating protein and is an important negative regulator for cell motility. Previous studies have demonstrated that DLC-1 functioned as a tumor suppressor gene and downregulation or even loss of DLC-1 expression often occurred in HCC [112]. The DLC-1 gene is potentially targeted by several differentially expressed miRNAs in HBc-introduced cells according to the miRNA-target gene network analysis. Thus, miR-382-5p seems to be significantly upregulated in HBc-overexpressing HCC cells. The HBc protein promotes HCC metastasis through enhancing the miR-382-5p level and reducing DLC-1 expression. The miR-382-5p/DLC-1 axis is essential for HBc-promoted HCC metastasis. These data further showed that, similar to HBx, HBc protein might also play multiple roles in different stages of HCC development [26]. A similar mechanism was observed with HCV. Indeed, miRNAs miR-141 and miR-200a are accentuated in HCV-infected human primary hepatocytes and can target DLC-1 mRNA reducing its expression and then induce HCC development [112].
A part of miRNA is closely related to the stage of liver disease. Indeed, studies have shown that the miRNA circulating in serum or plasma could serve as the role of biomarker for the diagnosis and prognosis of HBV-related diseases [113].
3.7. HBc Protein, a Regulator of Host Gene Expression
HBV targets host genes that are involved in cell survival to escape immune surveillance and facilitate malignant transformation. HBc protein may bind specifically to certain human gene promoters, through either its C-terminal functional domain or its N-terminal assembling domain. Previous research has generated the genome-wide profile of HBc in HBV-infected hepatocytes using chromatin immunoprecipitation microarray studies [20]. This study showed that HBc could bind to 64 gene promoters of the MAPK pathways and 41 gene promoters of the Wnt/β-catenin signaling pathways, whereas these two pathways are known to be critically involved in the development of HBV-related hepatocellular carcinoma. Moreover, the authors suggested that HBc tended to target the regulatory regions of genes with molecular function and malignant transformation in the liver cell repertoire. A previous study highlights that the accumulation of slight effects from HBc binding to many gene promoters may produce quite large effects on host cellular functions, possibly increasing a cell’s susceptibility to carcinogens [114].
Therefore, HBc has the ability to bind gene promoters in the human genome to modulate normal functions of liver cells infected with HBV and HBc could disrupt the expression of nearly 3100 human host genes by binding them to promoter regions [20].
3.8. Interaction with Viral Protein as HBx Protein
HBc protein seems to be able to interact with HBx, but the impact of this interaction on hepatocarcinogenesis seems to be controversial in some cases. Kwon et al., in their study of cultured HepG2 cells, showed that HBc and HBx proteins could synergistically repress both the promoter activity and the expression of the human p53 tumor suppressor gene [22]. The inactivation of the p53 gene participates in HCC progress and the synergistic action of these two proteins has an impact on malignant transformation.
In contrast, HBc and HBx proteins could reduce the expression of two Id proteins (Id1 and Id3) whereas Id proteins are supposed to be elevated in many tumor types and would correspond with the poor prognosis of HCC patients. Thus, HBc is capable of restraining the BMP/Smad signaling pathway and HBx is able to interact with both Id proteins for facilitating their degradation through proteasome-dependent manners. Therefore, it would be interesting to explore how HBV, one of the pathogenic factors of HCC, influences Id proteins [115].
Then, HBx protein can transactivate the expression of all HBV proteins through these two enhancers and can therefore increase the expression of the HBc in vitro and in vivo by transactivating the C promoter. The regulation of HBx level may be important in HCC development. In cultured human hepatoma cells, Kim et al. demonstrated that the level of HBx protein was significantly reduced by the co-expression of HBc, whereas the level of HBx mRNA was unaffected. The inhibitory effect of HBc is specific to HBx, and it did not affect other HBV proteins. It seems that HBx activates the synthesis of HBc during the early stage of viral replication and that HBc in turn functions as an effective downregulator of HBx. The regulation of the HBx level could involve the activation of the proteasome-mediated degradation of HBx. To date, no direct physical interaction between HBc and HBx has been demonstrated. Nevertheless, mutational analysis indicated that the C-terminal half of HBc is responsible for its inhibitory effect and that HBc protein controls the HBx level via a form of inhibitory feedback mechanism [116]. This study highlights a novel aspect of HBc function in the HBV life cycle and possibly in the development of HCC through control of the HBx level. To our knowledge, the molecular mechanism was not identified.
All of these elements emphasize a close link between HBc and HBx proteins, with a number of potential impacts on liver tumorigenesis.
4. Conclusions and Perspectives
Due to the high morbidity and mortality of HCC worldwide, for a number of years, many investigations on HCC carcinogenesis have been conducted that seek to elucidate the molecular mechanisms facilitating the design of better strategies to treat HCC. Evidence supports that the HBc protein has an oncogenic role in HBV-related HCC through several mechanisms, thereby controlling cancer cell proliferation and enabling malignant transformation. These include the signaling pathways involved in migration/proliferation of hepatoma cells; the resistance of cells to apoptosis; cell metabolic disorders; enhancing IL-6 expression and production; epigenetic alterations (miRNA); and other genetic processes.
With regard to garnering new insights into the biological roles of HBc in regulating HBV-related hepatocarcinogenesis, further exploration of the molecular mechanisms related to the dysfunction of hepatoma cells mediated by HBc may help us find novel therapeutic strategies for HBV-related HCC.
This entry is adapted from the peer-reviewed paper 10.3390/ijms222413651
References
- Liaw, Y.-F.; Chu, C.-M. Hepatitis B Virus Infection. Lancet 2009, 373, 582–592.
- Kim, B.K.; Han, K.-H.; Ahn, S.H. Prevention of Hepatocellular Carcinoma in Patients with Chronic Hepatitis B Virus Infection. Oncology 2011, 81 (Suppl. S1), 41–49.
- Ringelhan, M.; Protzer, U. Oncogenic Potential of Hepatitis B Virus Encoded Proteins. Curr. Opin. Virol. 2015, 14, 109–115.
- World Health Organization. Hepatitis B. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 9 November 2021).
- Summers, J.; Mason, W.S. Replication of the Genome of a Hepatitis B--like Virus by Reverse Transcription of an RNA Intermediate. Cell 1982, 29, 403–415.
- Guo, W.T.; Bell, K.D.; Ou, J.H. Characterization of the Hepatitis B Virus EnhI Enhancer and X Promoter Complex. J. Virol. 1991, 65, 6686–6692.
- Yee, J.K. A Liver-Specific Enhancer in the Core Promoter Region of Human Hepatitis B Virus. Science 1989, 246, 658–661.
- Chaturvedi, V.K.; Singh, A.; Dubey, S.K.; Hetta, H.F.; John, J.; Singh, M.P. Molecular Mechanistic Insight of Hepatitis B Virus Mediated Hepatocellular Carcinoma. Microb. Pathog. 2019, 128, 184–194.
- Parkin, D.M. The Global Health Burden of Infection-Associated Cancers in the Year 2002. Int. J. Cancer 2006, 118, 3030–3044.
- Kew, M.C. Epidemiology of Chronic Hepatitis B Virus Infection, Hepatocellular Carcinoma, and Hepatitis B Virus-Induced Hepatocellular Carcinoma. Pathol. Biol. 2010, 58, 273–277.
- Feitelson, M.A.; Lee, J. Hepatitis B Virus Integration, Fragile Sites, and Hepatocarcinogenesis. Cancer Lett. 2007, 252, 157–170.
- Farazi, P.A.; DePinho, R.A. Hepatocellular Carcinoma Pathogenesis: From Genes to Environment. Nat. Rev. Cancer 2006, 6, 674–687.
- Neuveut, C.; Wei, Y.; Buendia, M.A. Mechanisms of HBV-Related Hepatocarcinogenesis. J. Hepatol. 2010, 52, 594–604.
- Liu, W.; Guo, T.-F.; Jing, Z.-T.; Yang, Z.; Liu, L.; Yang, Y.-P.; Lin, X.; Tong, Q.-Y. Hepatitis B Virus Core Protein Promotes Hepatocarcinogenesis by Enhancing Src Expression and Activating the Src/PI3K/Akt Pathway. FASEB J. 2018, 32, 3033–3046.
- Bouchard, M.J.; Schneider, R.J. The Enigmatic X Gene of Hepatitis B Virus. J. Virol. 2004, 78, 12725–12734.
- Diab, A.; Foca, A.; Zoulim, F.; Durantel, D.; Andrisani, O. The Diverse Functions of the Hepatitis B Core/Capsid Protein (HBc) in the Viral Life Cycle: Implications for the Development of HBc-Targeting Antivirals. Antiviral. Res. 2018, 149, 211–220.
- Gai, X.; Zhao, P.; Pan, Y.; Shan, H.; Yue, X.; Du, J.; Zhang, Z.; Liu, P.; Ma, H.; Guo, M.; et al. Hepatitis B Virus Core Protein Enhances Human Telomerase Reverse Transcriptase Expression and Hepatocellular Carcinoma Cell Proliferation in a C-Ets2-Dependent Manner. Int. J. Biochem. Cell Biol. 2013, 45, 1174–1185.
- Kong, F.; Li, N.; Tu, T.; Tao, Y.; Bi, Y.; Yuan, D.; Zhang, N.; Yang, X.; Kong, D.; You, H.; et al. Hepatitis B Virus Core Protein Promotes the Expression of Neuraminidase 1 to Facilitate Hepatocarcinogenesis. Lab. Invest. 2020, 100, 1602–1617.
- Kong, F.; Tao, Y.; Yuan, D.; Zhang, N.; Li, Q.; Yu, T.; Yang, X.; Kong, D.; Ding, X.; Liu, X.; et al. Hepatitis B Virus Core Protein Mediates the Upregulation of C5α Receptor 1 via NF-ΚB Pathway to Facilitate the Growth and Migration of Hepatoma Cells. Cancer Res. Treat. 2020.
- Guo, Y.; Kang, W.; Lei, X.; Li, Y.; Xiang, A.; Liu, Y.; Zhao, J.; Zhang, J.; Yan, Z. Hepatitis B Viral Core Protein Disrupts Human Host Gene Expression by Binding to Promoter Regions. BMC Genom. 2012, 13, 563.
- Liu, W.; Lin, Y.-T.; Yan, X.-L.; Ding, Y.-L.; Wu, Y.-L.; Chen, W.-N.; Lin, X. Hepatitis B Virus Core Protein Inhibits Fas-Mediated Apoptosis of Hepatoma Cells via Regulation of MFas/FasL and SFas Expression. FASEB J. 2015, 29, 1113–1123.
- Kwon, J.A.; Rho, H.M. Transcriptional Repression of the Human P53 Gene by Hepatitis B Viral Core Protein (HBc) in Human Liver Cells. Biol. Chem. 2003, 384, 203–212.
- Du, J.; Liang, X.; Liu, Y.; Qu, Z.; Gao, L.; Han, L.; Liu, S.; Cui, M.; Shi, Y.; Zhang, Z.; et al. Hepatitis B Virus Core Protein Inhibits TRAIL-Induced Apoptosis of Hepatocytes by Blocking DR5 Expression. Cell Death Differ. 2009, 16, 219–229.
- Xie, Q.; Fan, F.; Wei, W.; Liu, Y.; Xu, Z.; Zhai, L.; Qi, Y.; Ye, B.; Zhang, Y.; Basu, S.; et al. Multi-Omics Analyses Reveal Metabolic Alterations Regulated by Hepatitis B Virus Core Protein in Hepatocellular Carcinoma Cells. Sci. Rep. 2017, 7, 41089.
- Chen, Z.; Li, Y.-X.; Fu, H.-J.; Ren, Y.-L.; Zou, L.; Shen, S.-Z.; Chen, P.; Sun, T.; Huang, C.-H. Hepatitis B Virus Core Antigen Stimulates IL-6 Expression via P38, ERK and NF-ΚB Pathways in Hepatocytes. Cell Physiol. Biochem. 2017, 41, 91–100.
- Du, J.; Bai, F.; Zhao, P.; Li, X.; Li, X.; Gao, L.; Ma, C.; Liang, X. Hepatitis B Core Protein Promotes Liver Cancer Metastasis through MiR-382-5p/DLC-1 Axis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1–11.
- Jeong, H.; Cho, M.-H.; Park, S.-G.; Jung, G. Interaction between Nucleophosmin and HBV Core Protein Increases HBV Capsid Assembly. FEBS Lett. 2014, 588, 851–858.
- Shim, H.Y.; Quan, X.; Yi, Y.-S.; Jung, G. Heat Shock Protein 90 Facilitates Formation of the HBV Capsid via Interacting with the HBV Core Protein Dimers. Virology 2011, 410, 161–169.
- Birnbaum, F.; Nassal, M. Hepatitis B Virus Nucleocapsid Assembly: Primary Structure Requirements in the Core Protein. J. Virol. 1990, 64, 3319–3330.
- Crowther, R.A.; Kiselev, N.A.; Böttcher, B.; Berriman, J.A.; Borisova, G.P.; Ose, V.; Pumpens, P. Three-Dimensional Structure of Hepatitis B Virus Core Particles Determined by Electron Cryomicroscopy. Cell 1994, 77, 943–950.
- Zheng, J.; Schödel, F.; Peterson, D.L. The Structure of Hepadnaviral Core Antigens. Identification of Free Thiols and Determination of the Disulfide Bonding Pattern. J. Biol. Chem. 1992, 267, 9422–9429.
- Blanchet, M.; Sureau, C. Analysis of the Cytosolic Domains of the Hepatitis B Virus Envelope Proteins for Their Function in Viral Particle Assembly and Infectivity. J. Virol. 2006, 80, 11935–11945.
- Sharma, R.R.; Dhiman, R.K.; Chawla, Y.; Vasistha, R.K. Immunohistochemistry for Core and Surface Antigens in Chronic Hepatitis. Trop. Gastroenterol. 2002, 23, 16–19.
- Petit, M.A.; Pillot, J. HBc and HBe Antigenicity and DNA-Binding Activity of Major Core Protein P22 in Hepatitis B Virus Core Particles Isolated from the Cytoplasm of Human Liver Cells. J. Virol. 1985, 53, 543–551.
- Duan, G.; Walther, D. The Roles of Post-Translational Modifications in the Context of Protein Interaction Networks. PLoS Comput. Biol. 2015, 11, e1004049.
- Lubyová, B.; Weber, J. Posttranslational Modifications of HBV Core Protein. Acta Virol. 2020, 64, 177–186.
- Ludgate, L.; Liu, K.; Luckenbaugh, L.; Streck, N.; Eng, S.; Voitenleitner, C.; Delaney, W.E.; Hu, J. Cell-Free Hepatitis B Virus Capsid Assembly Dependent on the Core Protein C-Terminal Domain and Regulated by Phosphorylation. J. Virol. 2016, 90, 5830–5844.
- Mondelli, M.; Tedder, R.S.; Ferns, B.; Pontisso, P.; Realdi, G.; Alberti, A. Differential Distribution of Hepatitis B Core and E Antigens in Hepatocytes: Analysis by Monoclonal Antibodies. Hepatology 1986, 6, 199–204.
- Hatton, T.; Zhou, S.; Standring, D.N. RNA- and DNA-Binding Activities in Hepatitis B Virus Capsid Protein: A Model for Their Roles in Viral Replication. J. Virol. 1992, 66, 5232–5241.
- Eckhardt, S.G.; Milich, D.R.; McLachlan, A. Hepatitis B Virus Core Antigen Has Two Nuclear Localization Sequences in the Arginine-Rich Carboxyl Terminus. J. Virol. 1991, 65, 575–582.
- Mak, L.-Y.; Wong, D.K.-H.; Seto, W.-K.; Lai, C.-L.; Yuen, M.F. Hepatitis B Core Protein as a Therapeutic Target. Expert. Opin. Ther. Targets 2017, 21, 1153–1159.
- Torresi, J.; Tran, B.M.; Christiansen, D.; Earnest-Silveira, L.; Schwab, R.H.M.; Vincan, E. HBV-Related Hepatocarcinogenesis: The Role of Signalling Pathways and Innovative Ex Vivo Research Models. BMC Cancer 2019, 19, 707.
- Haxho, F.; Neufeld, R.J.; Szewczuk, M.R. Neuraminidase-1: A Novel Therapeutic Target in Multistage Tumorigenesis. Oncotarget 2016, 7, 40860–40881.
- Glanz, V.Y.; Myasoedova, V.A.; Grechko, A.V.; Orekhov, A.N. Sialidase Activity in Human Pathologies. Eur. J. Pharmacol. 2019, 842, 345–350.
- Hou, G.; Liu, G.; Yang, Y.; Li, Y.; Yuan, S.; Zhao, L.; Wu, M.; Liu, L.; Zhou, W. Neuraminidase 1 (NEU1) Promotes Proliferation and Migration as a Diagnostic and Prognostic Biomarker of Hepatocellular Carcinoma. Oncotarget 2016, 7, 64957–64966.
- Amaddeo, G.; Cao, Q.; Ladeiro, Y.; Imbeaud, S.; Nault, J.-C.; Jaoui, D.; Gaston Mathe, Y.; Laurent, C.; Laurent, A.; Bioulac-Sage, P.; et al. Integration of Tumour and Viral Genomic Characterizations in HBV-Related Hepatocellular Carcinomas. Gut 2015, 64, 820–829.
- Wang, W.; Peng, J.X.; Yang, J.Q.; Yang, L.Y. Identification of Gene Expression Profiling in Hepatocellular Carcinoma Using CDNA Microarrays. Dig. Dis. Sci. 2009, 54, 2729–2735.
- Whittaker, S.; Marais, R.; Zhu, A.X. The Role of Signaling Pathways in the Development and Treatment of Hepatocellular Carcinoma. Oncogene 2010, 29, 4989–5005.
- Klein, N.P.; Schneider, R.J. Activation of Src Family Kinases by Hepatitis B Virus HBx Protein and Coupled Signaling to Ras. Mol. Cell Biol. 1997, 17, 6427–6436.
- Liu, H.; Xu, J.; Zhou, L.; Yun, X.; Chen, L.; Wang, S.; Sun, L.; Wen, Y.; Gu, J. Hepatitis B Virus Large Surface Antigen Promotes Liver Carcinogenesis by Activating the Src/PI3K/Akt Pathway. Cancer Res. 2011, 71, 7547–7557.
- Belgiovine, C.; Chiodi, I.; Mondello, C. Telomerase: Cellular Immortalization and Neoplastic Transformation. Multiple Functions of a Multifaceted Complex. Cytogenet. Genome Res. 2008, 122, 255–262.
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 1994, 266, 2011–2015.
- Hytiroglou, P.; Theise, N.D. Telomerase Activation in Human Hepatocarcinogenesis. Am. J. Gastroenterol. 2006, 101, 839–841.
- Giunco, S.; Dolcetti, R.; Keppel, S.; Celeghin, A.; Indraccolo, S.; Dal Col, J.; Mastorci, K.; De Rossi, A. HTERT Inhibition Triggers Epstein-Barr Virus Lytic Cycle and Apoptosis in Immortalized and Transformed B Cells: A Basis for New Therapies. Clin. Cancer Res. 2013, 19, 2036–2047.
- Nakayama, J.; Tahara, H.; Tahara, E.; Saito, M.; Ito, K.; Nakamura, H.; Nakanishi, T.; Tahara, E.; Ide, T.; Ishikawa, F. Telomerase Activation by HTRT in Human Normal Fibroblasts and Hepatocellular Carcinomas. Nat. Genet. 1998, 18, 65–68.
- Kojima, H.; Yokosuka, O.; Imazeki, F.; Saisho, H.; Omata, M. Telomerase Activity and Telomere Length in Hepatocellular Carcinoma and Chronic Liver Disease. Gastroenterology 1997, 112, 493–500.
- Tahara, H.; Nakanishi, T.; Kitamoto, M.; Nakashio, R.; Shay, J.W.; Tahara, E.; Kajiyama, G.; Ide, T. Telomerase Activity in Human Liver Tissues: Comparison between Chronic Liver Disease and Hepatocellular Carcinomas. Cancer Res. 1995, 55, 2734–2736.
- Kyo, S.; Takakura, M.; Fujiwara, T.; Inoue, M. Understanding and Exploiting HTERT Promoter Regulation for Diagnosis and Treatment of Human Cancers. Cancer Sci. 2008, 99, 1528–1538.
- Maida, Y.; Kyo, S.; Kanaya, T.; Wang, Z.; Yatabe, N.; Tanaka, M.; Nakamura, M.; Ohmichi, M.; Gotoh, N.; Murakami, S.; et al. Direct Activation of Telomerase by EGF through Ets-Mediated Transactivation of TERT via MAP Kinase Signaling Pathway. Oncogene 2002, 21, 4071–4079.
- Xu, D.; Dwyer, J.; Li, H.; Duan, W.; Liu, J.-P. Ets2 Maintains HTERT Gene Expression and Breast Cancer Cell Proliferation by Interacting with C-Myc. J. Biol. Chem. 2008, 283, 23567–23580.
- Barone, M.; Maiorano, E.; Ladisa, R.; Cuomo, R.; Pece, A.; Berloco, P.; Caruso, M.L.; Valentini, A.M.; Iolascon, A.; Francavilla, A.; et al. Influence of Ursodeoxycholate-Enriched Diet on Liver Tumor Growth in HBV Transgenic Mice. Hepatology 2003, 37, 880–886.
- Hsu, T.; Trojanowska, M.; Watson, D.K. Ets Proteins in Biological Control and Cancer. J. Cell Biochem. 2004, 91, 896–903.
- Sadik, C.D.; Miyabe, Y.; Sezin, T.; Luster, A.D. The Critical Role of C5a as an Initiator of Neutrophil-Mediated Autoimmune Inflammation of the Joint and Skin. Semin. Immunol. 2018, 37, 21–29.
- Woodruff, T.M.; Nandakumar, K.S.; Tedesco, F. Inhibiting the C5-C5a Receptor Axis. Mol. Immunol. 2011, 48, 1631–1642.
- Ajona, D.; Zandueta, C.; Corrales, L.; Moreno, H.; Pajares, M.J.; Ortiz-Espinosa, S.; Martínez-Terroba, E.; Perurena, N.; de Miguel, F.J.; Jantus-Lewintre, E.; et al. Blockade of the Complement C5a/C5aR1 Axis Impairs Lung Cancer Bone Metastasis by CXCL16-Mediated Effects. Am. J. Respir. Crit. Care Med. 2018, 197, 1164–1176.
- Kaida, T.; Nitta, H.; Kitano, Y.; Yamamura, K.; Arima, K.; Izumi, D.; Higashi, T.; Kurashige, J.; Imai, K.; Hayashi, H.; et al. C5a Receptor (CD88) Promotes Motility and Invasiveness of Gastric Cancer by Activating RhoA. Oncotarget 2016, 7, 84798–84809.
- Gu, J.; Ding, J.; Lu, C.; Lin, Z.; Chu, Y.; Zhao, G.; Guo, J.; Ge, D. Overexpression of CD88 Predicts Poor Prognosis in Non-Small-Cell Lung Cancer. Lung Cancer 2013, 81, 259–265.
- Hu, W.-H.; Hu, Z.; Shen, X.; Dong, L.-Y.; Zhou, W.-Z.; Yu, X.-X. C5a Receptor Enhances Hepatocellular Carcinoma Cell Invasiveness via Activating ERK1/2-Mediated Epithelial-Mesenchymal Transition. Exp. Mol. Pathol. 2016, 100, 101–108.
- Chen, C.; Wang, G. Mechanisms of Hepatocellular Carcinoma and Challenges and Opportunities for Molecular Targeted Therapy. World J. Hepatol. 2015, 7, 1964–1970.
- Ito, Y.; Sasaki, Y.; Horimoto, M.; Wada, S.; Tanaka, Y.; Kasahara, A.; Ueki, T.; Hirano, T.; Yamamoto, H.; Fujimoto, J.; et al. Activation of Mitogen-Activated Protein Kinases/Extracellular Signal-Regulated Kinases in Human Hepatocellular Carcinoma. Hepatology 1998, 27, 951–958.
- Moon, H.; Ro, S.W. MAPK/ERK Signaling Pathway in Hepatocellular Carcinoma. Cancers 2021, 13, 3026.
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome Classification of HCC Is Related to Gene Alterations and to New Therapeutic Targets. Hepatology 2007, 45, 42–52.
- Bortolami, M.; Kotsafti, A.; Cardin, R.; Farinati, F. Fas/FasL System, IL-1beta Expression and Apoptosis in Chronic HBV and HCV Liver Disease. J Viral Hepat 2008, 15, 515–522, doi:10.1111/j.1365-2893.2008.00974.x
- Kondo, T.; Suda, T.; Fukuyama, H.; Adachi, M.; Nagata, S. Essential Roles of the Fas Ligand in the Development of Hepatitis. Nat Med 1997, 3, 409–413, doi:10.1038/nm0497-409.
- Liang, X.; Liu, Y.; Zhang, Q.; Gao, L.; Han, L.; Ma, C.; Zhang, L.; Chen, Y.H.; Sun, W. Hepatitis B Virus Sensitizes Hepatocytes to TRAIL-Induced Apoptosis through Bax. J Immunol 2007, 178, 503–510, doi:10.4049/jimmunol.178.1.503.
- Hatano, E. Tumor Necrosis Factor Signaling in Hepatocyte Apoptosis. J Gastroenterol Hepatol 2007, 22 Suppl 1, S43-44, doi:10.1111/j.1440-1746.2006.04645.x.
- Müller, M.; Strand, S.; Hug, H.; Heinemann, E.M.; Walczak, H.; Hofmann, W.J.; Stremmel, W.; Krammer, P.H.; Galle, P.R. Drug-Induced Apoptosis in Hepatoma Cells Is Mediated by the CD95 (APO-1/Fas) Receptor/Ligand System and Involves Activation of Wild-Type P53. J Clin Invest 1997, 99, 403–413, doi:10.1172/JCI119174.
- Müller, M.; Scaffidi, C.A.; Galle, P.R.; Stremmel, W.; Krammer, P.H. The Role of P53 and the CD95 (APO-1/Fas) Death System in Chemotherapy-Induced Apoptosis. Eur Cytokine Netw 1998, 9, 685–686.
- Hsu, I.C.; Metcalf, R.A.; Sun, T.; Welsh, J.A.; Wang, N.J.; Harris, C.C. Mutational Hotspot in the P53 Gene in Human Hepato-cellular Carcinomas. Nature 1991, 350, 427–428, doi:10.1038/350427a0.
- Aguilar, F.; Harris, C.C.; Sun, T.; Hollstein, M.; Cerutti, P. Geographic Variation of P53 Mutational Profile in Nonmalignant Human Liver. Science 1994, 264, 1317–1319, doi:10.1126/science.8191284.
- Dockrell, D.H. The Multiple Roles of Fas Ligand in the Pathogenesis of Infectious Diseases. Clin Microbiol Infect 2003, 9, 766–779, doi:10.1046/j.1469-0691.2003.00669.x.
- Johnson, D.G.; Schneider-Broussard, R. Role of E2F in Cell Cycle Control and Cancer. Front Biosci 1998, 3, d447-448, doi:10.2741/a291.
- Yamasaki, L.; Jacks, T.; Bronson, R.; Goillot, E.; Harlow, E.; Dyson, N.J. Tumor Induction and Tissue Atrophy in Mice Lacking E2F-1. Cell 1996, 85, 537–548, doi:10.1016/s0092-8674(00)81254-4.
- Pierce, A.M.; Schneider-Broussard, R.; Gimenez-Conti, I.B.; Russell, J.L.; Conti, C.J.; Johnson, D.G. E2F1 Has Both Oncogenic and Tumor-Suppressive Properties in a Transgenic Model. Mol Cell Biol 1999, 19, 6408–6414, doi:10.1128/mcb.19.9.6408.
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A. Identification and Characterization of a New Member of the TNF Family That Induces Apoptosis. Immunity 1995, 3, 673–682, doi:10.1016/1074-7613(95)90057-8.
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.; Gentz, R.; Dixit, V.M. An Antagonist Decoy Receptor and a Death Domain-Containing Re-ceptor for TRAIL. Science 1997, 277, 815–818, doi:10.1126/science.277.5327.815.
- Lagunas-Martínez, A.; Madrid-Marina, V.; Gariglio, P. Modulation of Apoptosis by Early Human Papillomavirus Proteins in Cervical Cancer. Biochim Biophys Acta 2010, 1805, 6–16, doi:10.1016/j.bbcan.2009.03.005.
- Liang, X.; Du, J.; Liu, Y.; Cui, M.; Ma, C.; Han, L.; Qu, Z.; Zhang, Z.; Sun, Z.; Zhang, L.; et al. The Hepatitis B Virus Protein MHBs(t) Sensitizes Hepatoma Cells to TRAIL-Induced Apoptosis through ERK2. Apoptosis 2007, 12, 1827–1836, doi:10.1007/s10495-007-0114-4.
- Liu, Y.-G.; Liu, S.-X.; Liang, X.-H.; Zhang, Q.; Gao, L.-F.; Han, L.-H.; Cao, Y.-L.; Hou, N.; Du, J.; Sun, W.-S. Blockade of TRAIL Pathway Ameliorates HBV-Induced Hepatocyte Apoptosis in an Acute Hepatitis Model. Biochem Biophys Res Commun 2007, 352, 329–334, doi:10.1016/j.bbrc.2006.11.024.
- Yoshida, T.; Maeda, A.; Tani, N.; Sakai, T. Promoter Structure and Transcription Initiation Sites of the Human Death Receptor 5/TRAIL-R2 Gene. FEBS Lett 2001, 507, 381–385, doi:10.1016/s0014-5793(01)02947-7
- Jia, B.; Guo, M.; Li, G.; Yu, D.; Zhang, X.; Lan, K.; Deng, Q. Hepatitis B Virus Core Protein Sensitizes Hepatocytes to Tumor Necrosis Factor-Induced Apoptosis by Suppression of the Phosphorylation of Mitogen-Activated Protein Kinase Kinase 7. J. Virol. 2015, 89, 2041–2051, doi:10.1128/JVI.03106-14.
- Fritz, V.; Fajas, L. Metabolism and Proliferation Share Common Regulatory Pathways in Cancer Cells. Oncogene 2010, 29, 4369–4377, doi:10.1038/onc.2010.182.
- Lévy, P.; Bartosch, B. Metabolic Reprogramming: A Hallmark of Viral Oncogenesis. Oncogene 2016, 35, 4155–4164, doi:10.1038/onc.2015.479.
- Ringelhan, M.; Heikenwalder, M.; Protzer, U. Direct Effects of Hepatitis B Virus-Encoded Proteins and Chronic Infection in Liver Cancer Development. Dig Dis 2013, 31, 138–151, doi:10.1159/000347209.
- You, X.; Liu, F.; Zhang, T.; Li, Y.; Ye, L.; Zhang, X. Hepatitis B Virus X Protein Upregulates Oncogene Rab18 to Result in the Dysregulation of Lipogenesis and Proliferation of Hepatoma Cells. Carcinogenesis 2013, 34, 1644–1652, doi:10.1093/carcin/bgt089.
- Li, X.; Jiang, Y.; Meisenhelder, J.; Yang, W.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Aldape, K.; He, J.; Hunter, T.; et al. Mitochon-dria-Translocated PGK1 Functions as a Protein Kinase to Coordinate Glycolysis and the TCA Cycle in Tumorigenesis. Mol Cell 2016, 61, 705–719, doi:10.1016/j.molcel.2016.02.009.
- Wang, Y.; Wu, T.; Hu, D.; Weng, X.; Wang, X.; Chen, P.-J.; Luo, X.; Wang, H.; Ning, Q. Intracellular Hepatitis B Virus Increases Hepatic Cholesterol Deposition in Alcoholic Fatty Liver via Hepatitis B Core Protein. J Lipid Res 2018, 59, 58–68, doi:10.1194/jlr.M079533.
- Ohishi, W.; Cologne, J.B.; Fujiwara, S.; Suzuki, G.; Hayashi, T.; Niwa, Y.; Akahoshi, M.; Ueda, K.; Tsuge, M.; Chayama, K. Serum Interleukin-6 Associated with Hepatocellular Carcinoma Risk: A Nested Case-Control Study. Int J Cancer 2014, 134, 154–163, doi:10.1002/ijc.28337.
- Taniguchi, K.; Karin, M. IL-6 and Related Cytokines as the Critical Lynchpins between Inflammation and Cancer. Semin Immunol 2014, 26, 54–74, doi:10.1016/j.smim.2014.01.001.
- Xia, C.; Liu, Y.; Chen, Z.; Zheng, M. Involvement of Interleukin 6 in Hepatitis B Viral Infection. Cell Physiol Biochem 2015, 37, 677–686, doi:10.1159/000430386.
- Nakagawa, H.; Maeda, S.; Yoshida, H.; Tateishi, R.; Masuzaki, R.; Ohki, T.; Hayakawa, Y.; Kinoshita, H.; Yamakado, M.; Kato, N.; et al. Serum IL-6 Levels and the Risk for Hepatocarcinogenesis in Chronic Hepatitis C Patients: An Analysis Based on Gender Differences. Int J Cancer 2009, 125, 2264–2269, doi:10.1002/ijc.24720.
- Sugimoto, Y.; Wakai, K.; Nakagawa, H.; Suma, S.; Sasakabe, T.; Sakamoto, T.; Takashima, N.; Suzuki, S.; Ogawa, S.; Ohnaka, K.; et al. Associations between Polymorphisms of Interleukin-6 and Related Cytokine Genes and Serum Liver Damage Markers: A Cross-Sectional Study in the Japan Multi-Institutional Collaborative Cohort (J-MICC) Study. Gene 2015, 557, 158–162, doi:10.1016/j.gene.2014.12.025.
- Jang, J.W.; Oh, B.S.; Kwon, J.H.; You, C.R.; Chung, K.W.; Kay, C.S.; Jung, H.S. Serum Interleukin-6 and C-Reactive Protein as a Prognostic Indicator in Hepatocellular Carcinoma. Cytokine 2012, 60, 686–693, doi:10.1016/j.cyto.2012.07.017.
- Lee, Y.; Park, U.S.; Choi, I.; Yoon, S.K.; Park, Y.M.; Lee, Y.I. Human Interleukin 6 Gene Is Activated by Hepatitis B Virus-X Protein in Human Hepatoma Cells. Clin Cancer Res 1998, 4, 1711–1717.
- Kim, J.S.; Rho, B. young; Lee, T.H.; Lee, J.M.; Kim, S.J.; Park, J.H. The Interaction of Hepatitis B Virus X Protein and Protein Phosphatase Type 2 Calpha and Its Effect on IL-6. Biochem Biophys Res Commun 2006, 351, 253–258, doi:10.1016/j.bbrc.2006.10.028.
- Xiang, W.-Q.; Feng, W.-F.; Ke, W.; Sun, Z.; Chen, Z.; Liu, W. Hepatitis B Virus X Protein Stimulates IL-6 Expression in Hepatocytes via a MyD88-Dependent Pathway. J Hepatol 2011, 54, 26–33, doi:10.1016/j.jhep.2010.08.006.
- Tarocchi, M.; Polvani, S.; Marroncini, G.; Galli, A. Molecular Mechanism of Hepatitis B Virus-Induced Hepatocarcinogenesis. World J Gastroenterol 2014, 20, 11630–11640, doi:10.3748/wjg.v20.i33.11630.
- Bushati, N.; Cohen, S.M. MicroRNA Functions. Annu Rev Cell Dev Biol 2007, 23, 175–205, doi:10.1146/annurev.cellbio.23.090506.123406.
- Scaria, V.; Hariharan, M.; Maiti, S.; Pillai, B.; Brahmachari, S.K. Host-Virus Interaction: A New Role for MicroRNAs. Retrovi-rology 2006, 3, 68, doi:10.1186/1742-4690-3-68.
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. MiR-122, a Mammalian Liver-Specific MicroRNA, Is Processed from Hcr MRNA and May Downregulate the High Affinity Cationic Amino Acid Transporter CAT-1. RNA Biol 2004, 1, 106–113, doi:10.4161/rna.1.2.1066.
- Loureiro, D.; Tout, I.; Narguet, S.; Benazzouz, S.M.; Mansouri, A.; Asselah, T. MiRNAs as Potential Biomarkers for Viral Hepatitis B and C. Viruses 2020, 12, E1440, doi:10.3390/v12121440.
- Banaudha, K.; Kaliszewski, M.; Korolnek, T.; Florea, L.; Yeung, M.L.; Jeang, K.-T.; Kumar, A. MicroRNA Silencing of Tumor Suppressor DLC-1 Promotes Efficient Hepatitis C Virus Replication in Primary Human Hepatocytes. Hepatology 2011, 53, 53–61, doi:10.1002/hep.24016.
- Wei, Y.-F.; Cui, G.-Y.; Ye, P.; Chen, J.-N.; Diao, H.-Y. MicroRNAs May Solve the Mystery of Chronic Hepatitis B Virus Infec-tion. World J Gastroenterol 2013, 19, 4867–4876, doi:10.3748/wjg.v19.i30.4867.
- Zheng, Y.; Chen, W.; Louie, S.G.; Yen, T.S.B.; Ou, J.J. Hepatitis B Virus Promotes Hepatocarcinogenesis in Transgenic Mice. Hepatology 2007, 45, 16–21, doi:10.1002/hep.21445.
- Xia, L.; Wang, S.; Zhang, H.; Yang, Y.; Wei, J.; Shi, Y.; Zou, C.; Liu, J.; Luo, M.; Huang, A.; et al. The HBx and HBc of Hepatitis B Virus Can Influence Id1 and Id3 by Reducing Their Transcription and Stability. Virus Res 2020, 284, 197973, doi:10.1016/j.virusres.2020.197973.
- Kim, J.-H.; Kang, S.; Kim, J.; Ahn, B.-Y. Hepatitis B Virus Core Protein Stimulates the Proteasome-Mediated Degradation of Viral X Protein. J Virol 2003, 77, 7166–7173, doi:10.1128/jvi.77.13.7166-7173.2003.