Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemical Research Methods
1,3-Dienes are vital building blocks in organic synthesis. They underpin many fundamental synthetic transformations and are present in numerous natural products and drug candidate molecules.
- allene
- rearrangement
- stereoselective
- transition metal
1. Introduction
1,3-Dienes (1) are essential building blocks in organic synthesis and materials chemistry. They have been crucial in the development of fundamental synthetic reactions such as the Diels-Alder and related pericyclic transformations [1,2,3,4,5]. The 1,3-diene (1) structural motif can be found across numerous natural product classes, as well as in several high-profile on-market drugs and drug candidates; their inherent electronic characteristics have made them indispensable in the development of innovative photoactive organic materials [6,7,8,9]. Given their significance, the synthesis of 1,3-dienes (1) is rich in history and innovation, and a recent extensive review, centered on stereoselective methods provides an excellent account of current synthetic approaches to the 1,3-diene motif [10]. The application of olefination strategies has dominated their synthesis [11,12,13,14,15,16]; this was subsequently augmented by transition metal cross-coupling approaches [17,18,19,20,21], and the bond reorganization of appropriately substituted alkene and alkyne substrates [22,23,24,25], giving rise to 1,3-dienes (1) with unique connectivity’s that can be tailored to the synthetic target of choice (Scheme 1).
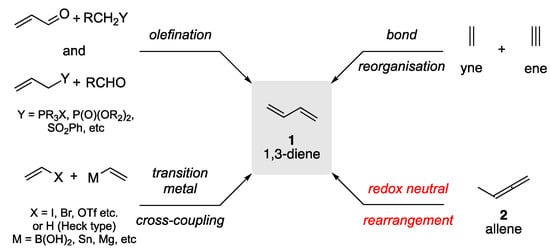
Scheme 1. Synthesis of 1,3-dienes (1).
The rearrangement of an alkyl-allene (2) is an appealing approach to the synthesis 1,3-dienes 1, as it is redox neutral and can be viewed as a formal 1,3-hydrogen migratory process (Scheme 2). Unlike olefination strategies, transition metal cross-coupling, and bond-reorganization approaches, this approach can utilize the inherent thermodynamic stability of the product 1,3-diene to promote the rearrangement.
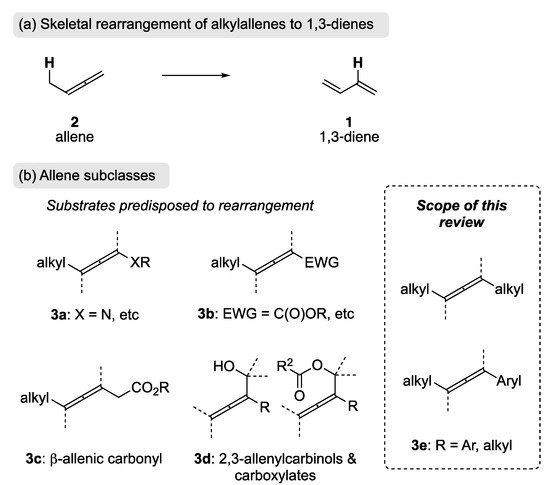
Scheme 2. (a) Rearrangement of an alkylallene to a 1,3-diene. (b) Different allene subclasses.
The rearrangement of allenes activated by a heteroatom attached directly to the allene (3a) has been reported by Hsung et al. [26,27], and an extensive review has been published by the same authors [28]. An electron-withdrawing group attached directly to the allene will bias it toward rearrangement (e.g., 3b); this has been realized through phosphine-catalyzed conjugate addition [29,30,31] and via palladium catalysis [32]. β-Allenic carbonyl substrates (3c) are also predisposed to rearrangement, and these substrates can be readily re-conjugated via base catalysis thereby providing dienoic esters, which are present in several natural occurring pheromones and flavors [33,34,35]. Additionally, while the rearrangement of 2,3-allenylcarbinols and their corresponding carboxylates (e.g., 3d) can be achieved through a [3,3]-sigmatropic rearrangement [36,37,38], SN2′ pathways [39,40,41], and metal-mediated processes [42,43,44], it does not fall within the scope of this review, as it is not a formal 1,3-hydrogen migratory process. With the emergence of allenes as building blocks in organic synthesis [45,46], the rearrangement of 2 to 1 (Scheme 2) has the potential to be a valuable, complementary tool, in the synthesis of 1,3-dienes. The focus of this review will therefore be the rearrangement of allenes not predisposed to rearrangement through direct attachment of heteroatoms or electron-withdrawing groups (e.g., 3e).
2. Acid-Mediated Rearrangements
Acid-mediated rearrangements are primarily realized through the protonation of the central sp-hybridized carbon of the allene, where stabilization of a transient carbocation facilitates the formation of a thermodynamically stable 1,3-diene. This method, therefore, limits the scope of the allene substrates that be effectively rearranged to their 1,3-diene counterparts.
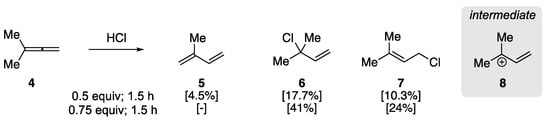
In 1960, Johnson et al. examined the addition of hydrogen chloride to aliphatic allenes to elucidate the regiochemistry of the initial protonation [47]. This early report observed the rearrangement of 3-methyl-1,2-butadiene (4) to isoprene in the presence of HCl (Scheme 3), clarifying previous studies on the addition of HX to allenes. They found that the treatment of 4 with HCl (0.5 equiv) at −78 °C for 0.5 h yielded a mixture of isoprene 5 and allylic chloride addition products (6/7), as determined by GC analysis. The ratio of 5 to 6/7 could be affected through HCl stoichiometry and reaction time, where an increase in the amount of HCl and reaction time resulted in primarily allylic chlorides 6 and 7, respectively. The authors rationalized the formation of the products by the protonation of the central sp-hybridized carbon yielding carbocation 8, with the formation of 1,3-diene 5 resulting from subsequent elimination.
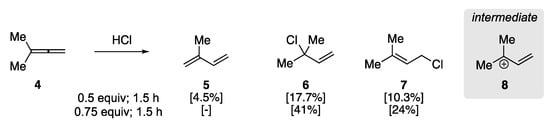
Scheme 3. Acid-catalyzed rearrangement of 3-methyl-1,2-butadiene 4 [47].
Wenkert et al. found that allenes 9 and 11 could be rearranged to 1,3-dienes 10 and 12, respectively (Scheme 4). [48]. Analogous to the work of Johnson, protonation occurs through the central sp-hybridized allenic carbon to provide an intermediate such as 13, which can be further stabilized by the electron-donating 4-methoxy aryl substituent. Consequently, both allenes 9 and 11 can be considered as being activated.
Scheme 4. HCl-promoted allene rearrangement. The rearrangement was facilitated by the presence of an electron-rich 4MeO-arene substituent [48].
Kropp et al. had demonstrated that silica gel and alumina facilitated the addition of HX to alkenes and alkynes [49]. In a related study, they observed that tetramethylallene 14 could be rearranged to 2,4-dimethylpenta-1,3-diene (15) through treatment with silica gel (Scheme 5). However, they found that extended reactions times (24 h) were required to achieve modest conversion and that 15 would undergo further dimerization to produce 16.
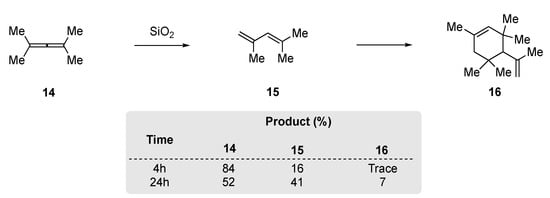
Scheme 5. SiO2 rearrangement of tetramethylallene 14 [49].
Li et al. utilized the intrinsic strain of diarylvinylidenecyclopropanes in their acid mediate rearrangement to yield trienes (Scheme 6) [50]. For example, the dicyclopentyl (17a) and dicyclohexyl (17b) diarylvinylidenecyclopropanes could be effectively rearranged to provide trienes 18a and 18b, in 65% and 50% isolated yield, respectively. Additionally, using DFT calculations they identified the formation of the carbocation 19 as key within this rearrangement.
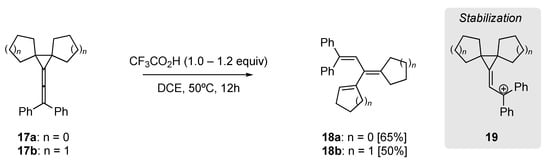
Scheme 6. Trifluoroacetic acid-mediated rearrangement of diaryl-vinylidenecyclopropanes [50].
In 2010, Sanz et al. treated indolyl allenes (20) in refluxing MeCN and catalytic PTSA, yielding 1,3-dienes 21 in high isolated yield (Scheme 7) [51]. The rearrangement of this allene subgroup benefited from subsequent stabilization of carbocation 22, formed upon protonation with PTSA. Stabilization is afforded through the participation of electron-rich indolyl substituent, as well as the flanking phenyl groups, although there appears to be no perceived benefit in isolated chemical yield when using electron-poor (20b, Ar = 4F-C6H4) or electron-rich (20c, Ar = 4MeOC6H4) aryl substituents.
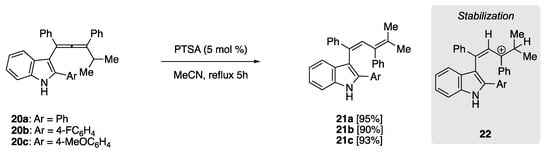
Scheme 7. PTSA-catalyzed rearrangement of indolylallenes to yield penta-substituted 1,3-dienes. The intermediate tertiary carbocation is stabilized by the electron-rich indolyl substituent [51].
Titov et al., in a study examining the synthesis of potential bioactive tetrahydrobenzazecines, observed that cyclic allene 23 underwent rearrangement to 1,3-diene 24 upon microwave irradiation in acetic acid (Scheme 8) [52]. The yields were found to be good to excellent, depending on the substitution. Again, this type of rearrangement was believed to occur by the benzylic carbocation 25. This intermediate also benefits from stabilization via the N-heteroatom of the enamine. Another key driving force for the formation of diene 24, could well be the release of the inherent ring strain within cyclic allene 23.
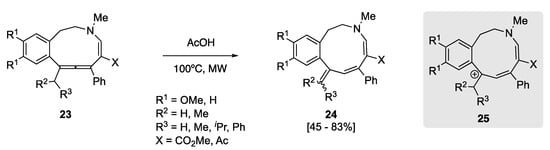
Scheme 8. Acetic acid- and microwave-mediated rearrangement of allenes 23 [52].
3. Thermal-Mediated Rearrangements
The rearrangement of strained allenic systems, such as the cyclopropyl allenes 26 and 28, to 1,3-dienes has been known since the late 1960s (Scheme 9). Seminal works by Crandall and Paulson [53], Conia et al. [54], and Maitland Jones et al. [55] demonstrated that thermal rearrangements of strained exo-cyclic allenyl cyclopropanes, employing pyrolysis through a flow system at temperatures in excess of 300 °C, gave access to hitherto unknown 1,3-dienyl cyclopropanes through a homolytic ring breakage and subsequent ring-closure events.
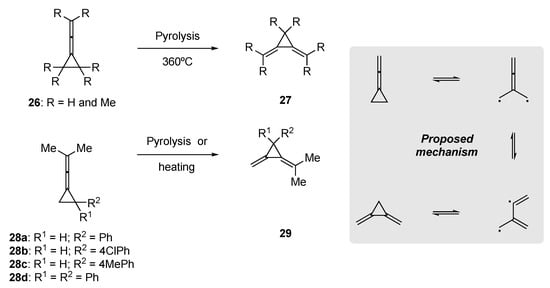
The synthetic utility of this transformation is limited. Rearrangement in these strained allenic systems (e.g., 28a–d) benefits from aryl substitution on the cyclopropane, and this aligns well with the proposed mechanism to provide added stabilization in the radical process.
In 1968, Taylor and Wright re-examined the thermal rearrangement of tetramethylallene 14, as prior reports had shown that 14 undergoes dimerization at 150 °C [56]. They observed that heating of 14 in a glass vessel that had been pre-treated with base gave the dimer, tetramethyl-1,2-di-isopropylidenecyclobutane. However, in an unwashed or acid-treated vessel, rapid rearrangement to 1,3-diene, 2,4-dimethylpenta-1,3-diene (15) was observed; subsequently, 15 underwent further dimerization to yield a complex mixture. This led to a synthetically useful procedure being established, where the allene 14 could be successfully rearranged to 2,4-dimethylpenta-1,3-diene (15) through heating a diluted solution in a polar aprotic solvent at elevated temperatures (Scheme 10).

Scheme 10. Thermal rearrangement of tetramethylallene in polar aprotic solvents [56].
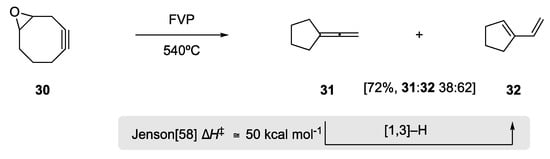
In 1989, Meier and Schmitt observed a formal 1,3-hydrogen shift in the exo-cyclic allene 31 (Scheme 11) [57]. Upon Flash Vacuum Pyrolysis of the cyclooctynyl epoxide 30, they observed two major products in 72% yield, exo-cyclic allene 31 and 1,3-diene 32 in a 38:62 ratio. The 1,3-diene was postulated to occur through formal [1,3]-hydrogen migration. Experimentally this reaction occurs at 540 °C under the FVP conditions; Jenson, in a very insightful theoretical study on the effects of allenes in sigmatropic H shift, subsequently calculated an energy barrier of ΔH‡ of ≅50 kcal mol−1 for this thermal [1,3]-H rearrangement [58].
This entry is adapted from the peer-reviewed paper 10.3390/reactions3010006
This entry is offline, you can click here to edit this entry!