Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Agriculture, Dairy & Animal Science
p53, a critical tumor suppressor, is commonly mutated in neoplasia, including colorectal cancer. To devise anti-cancer strategies targeting p53, it is crucial to understand the myriad cell-specific regulatory mechanisms in the p53 signaling pathway, and how these same regulatory mechanisms may be evaded by p53 mutants.
- degradation
- colorectal cancer
- p53
- MDM2
- negative feedback
1. Regulation of Gain-of-Function p53 Mutant Expression
Loss of p53 function is a hallmark of cancer development, specifically in the CRC adenoma-to-carcinoma transition. Many p53 mutations result in loss of functional proteins (74–80%) [72], but some result in proteins with gain of function (GOF), i.e., with novel activities [73,74].
The importance of GOF phenotypes first became apparent when mutp53 expression in a p53-null cell line displayed oncogenic properties [75]. Later, individuals with germline p53 missense mutations (Li–Fraumeni Syndrome) were found to have earlier cancer onset and shorter survival compared to those with germline p53 null mutants [76,77]; these findings were supported by similar observations in animal models [78]. Oncogenic phenotypes can be abolished if mutp53 expression is suppressed or ablated in vivo [79]. These GOF mutations enhance cell proliferation, increase colony formation, and promote cell invasion and migration, angiogenesis, and chromatin remodeling.
GOF mutants often arise from accumulation of missense mutations in tumor cells [80,81,82]. Malignant transformation is believed to be triggered by DNA damage, at least in the R175 mutants [80]. Similar to wild-type p53, MDM2 appears capable of regulating mutp53, as evidenced by enhanced mutp53 levels when mutp53-bearing animals are crossed with MDM2 KO mice [82]. The assumption is that mutp53 cannot trigger the same downstream pathway as wild-type p53, including induction of MDM2; consequently, low MDM2 abundance results in increased mutp53 stability. Indeed, mutp53 mice subjected to ROS, radiation, and DNA-damaging chemotherapy stabilized mutp53 protein, resulting in earlier tumor onset and shorter survival [83]. A recent report demonstrates that some mutp53s can bind to the central zinc finger/acidic domain of MDM2, thereby directly inhibiting its E3 ligase activity—another mechanism prolonging mutp53 half-life [84].
Although p53 GOF mutants have survival advantages, loss of heterozygosity (LOH) permits GOF mutants to reach their full malignant potential. Approximately 93% of human cancers lose wild-type p53 through loss of heterozygosity [85], and its mechanistic importance was best seen in stabilizing nuclear localization of mutp53 [85,86,87]. Using a R270H mutant in murine intestinal organoid models, Nakayama et al. demonstrated that wild-type p53 suppressed the mutp53 phenotype; LOH enhances the metastatic capability of tumors by increasing mutp53 stability and nuclear localization [88].
Studies tend to generalize mutp53s, but they are functionally diverse and could have varying prognostic values. In a compilation of survival analysis of several cancer types, including CRC, from a combined dataset of The Cancer Genome Atlas (TCGA) and MSKCC bladder cancer dataset (JCO, 2013), totaling 2916 cases, Xu et al. [89] examined how individual p53 hotspot mutations affect outcomes in human cancer. Survival outcomes appear similar between structural, contact, and nonsense mutations. Surprisingly, survival outcome also did not differ significantly between wild-type p53 and mutp53. When the survival outcomes of individual GOF mutations were compared to nonsense mutation using Kaplan–Meier survival analyses, patients with R248 and R282 mutations-bearing tumors had a significantly worse prognosis. These findings were validated in a separate dataset. To identify the mechanism contributing to the worse prognosis associated with R248 and R282 mutations, Xu et al. performed gene enrichment analysis on the CRC dataset in TCGA (n = 224 [3]). They showed that, unlike the two intensely studied p53 mutations, R175H and R273H, R248Q/W and R282W mutations shared 52 commonly enriched genes: The shared pathways center around cytochrome p450 CYP3A4, an important gene for drug metabolism. CYP3A4 expressions were elevated in tumors bearing R248Q/W and R282W p53 mutations; enhanced tumor resistance to chemotherapy is likely to explain worse survival outcomes.
2. Therapeutic Strategies against p53 Mutants
Tumor dependence (addiction) on mutp53 GOF led to targeted therapy to suppress mutp53 expression by protein degradation (Figure 2), transcriptional suppression, and conversion to wild-type p53 phenotype. Novel immunotherapies leveraging neoantigens in mutp53 are also emerging. These approaches are discussed below.
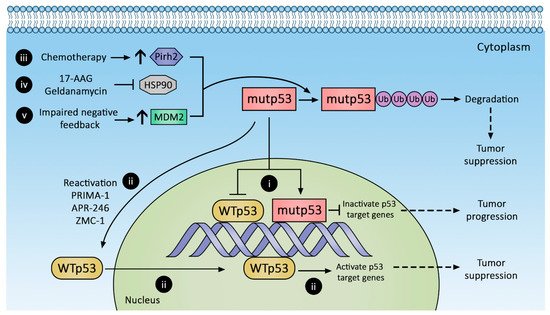
Figure 2. Therapeutic strategies against p53 mutants (mutp53). Mutp53 promotes CRC progression, whereas reactivation and decreased mutp53 suppress CRC. Mutp53 inactivation of p53 target genes (i) can be reversed by (ii) reactivating wild-type p53 with PRIMA-1, APR-246, and ZMC-1 or augmenting mutp53 degradation with (iii) chemotherapy to increase Pirh2 expression, (iv) inhibiting HSP90, and (v) increasing MDM2 expression.
2.1. Promoting mutp53 Protein Degradation
2.1.1. Increasing MDM2 Expression
As reviewed above, mutp53 accumulation has been attributed to impaired negative feedback between MDM2 and mutp53. MDM2 can bind and ubiquitinate mutp53, although with decreased efficiency compared to wild-type p53 [90,91]. Augmenting MDM2 expression is believed to degrade mutp53 and suppress tumor growth, but a recent study by Yang et al. revealed that mutp53 (R175H, G245S, R248Q, R273H, D281G) inhibits MDM2 activity [84]. In addition, desired depletion of mutp53 in tumors or generalized depletion of wild-type p53 in normal tissues are difficult to achieve. The other concern is that a global increase in MDM2 expression may have an untoward effect on normal tissues and inadvertently promote tumorigenesis. The feasibility of this strategy has yet to be tested in the clinic.
2.1.2. Stabilizing MDM2-mutp53 Complex
Theoretically, small molecules can be designed to target the abovementioned MDM2 modulators via an agonist or antagonist mechanism. However, few, if any, are in preclinical testing, and to our knowledge, none have been evaluated for colorectal cancer.
2.1.3. Hsp90 Inhibition
Mutp53 subtypes exploit protein folding machinery such as Hsp90 to increase their half-life, and the inadvertent activation of stress responses to misfolded proteins also enhances malignant cell survival. Of the six p53 mutation “hot spots” (R175, G245, R248, R249, R273, R282), nearly all demonstrate prolonged engagement with the Hsp90 complex compared to wild-type p53, suggesting they are prone to protein misfolding [50]. Geldanamycin (Figure 2(iv)) is a first-generation Hsp90 inhibitor that shows great efficacy in diverting p53 conformation mutants toward the Hsp70-CHIP complex to promote their degradation [48]. However, despite antitumor efficacy, geldanamycin is plagued with adverse effects, most notably hepatotoxicity [92]. Geldanamycin analogues with reduced toxicity have been engineered (e.g., 17-AAG), but they have low oral bioavailability [93].
Newer generations of non-geldanamycin-derived Hsp90 inhibitors emerged in the last decade. Ganetespib, an early agent in this class, is 50-fold more potent than 17-AAG at inhibiting Hsp90 [79], with improved solubility and reduced toxicity [94]. TAS-116, another novel agent, is orally active and selective against cytoplasmic Hsp90. Although TAS-116 monotherapy shows limited antitumor effect [95], TAS-116 plus nivolumab, an immune checkpoint inhibitor, demonstrates an augmented treatment response against microsatellite-stable (MSS) CRC compared to TAS-116 monotherapy [96].
HDAC6 regulates Hsp90 expression and its associated protein machinery; inhibition of HDAC6 hyperacetylates Hsp90, thereby abolishing Hsp90 binding to its partners [97]. SAHA, a HDAC inhibitor explored as an alternative method of inhibiting Hsp90, demonstrates preferential toxicity to mutp53-expressing cells [98].
2.1.4. Pirh2 Upregulation
Another approach to promote mutp53 degradation is with arsenic trioxide, which induces Pirh2 expression and promotes proteasomal degradation of mutp53s [99,100] (Figure 2(iii)). Mutp53s susceptible to Pirh2-mediated regulation include R175H, R248W, H179Y/R282W, and R273H. Arsenic trioxide is commonly evaluated as part of combination therapy, e.g., with Gemtuzumab ozogamicin in previously untreated acute promyelocytic leukemia (NCT01409161) or with itraconazole in advanced basal cell cancer (NCT02699723). In 2010, a phase 1 trial of 5-fluorouracil, leucovorin, and arsenic trioxide for refractory CRC established a safe dose [101], but no phase 2 trial is currently registered on clinicaltrials.gov.
2.2. Conversion of mutp53 to Wild-Type p53
Selective secondary mutations in the C-terminal domain of mutp53 can restore proper protein folding in the transactivation domain and DNA-binding domains, thereby restoring wild-type p53 characteristics [102,103,104]. Small synthetic peptides with similar allosteric stabilization properties targeting the C-terminal domain of mutp53 can also restore the wild-type p53 phenotype. A search for small molecules with the abovementioned properties yielded PRIMA-1 and its analog PRIMA-1Met (also known as APR-246). PRIMA-1 and APR-246 (Figure 2(ii)) are prodrugs of the active compound methylene quinuclidinone (MQ), which forms covalent bonds with cysteines in mutp53, restoring wild-type-like sequence-specific binding and inducing tumor apoptosis [105,106,107]. Their clinical efficacy remains unknown. A highly anticipated phase 1b/2 clinical trial combining APR-246 with carboplatin for the treatment of recurrent high-grade ovarian cancer (NCT02098343) was completed in April 2019, but no results have been reported. Other trials evaluating combined APR-246 and azacitadine for various hematologic malignancies (NCT03931291, NCT04214860, NCT03745716, NCT03588078) are ongoing, with at least one in phase 3.
Zinc metalochepaeron-1 (ZMC-1; NSC319726), another compound capable of reactivating mutp53 (Figure 2(ii)), was discovered in a drug screen using an NCI60 tumor cell line panel [108]. ZMC-1 promotes zinc binding with mutp53, thereby stabilizing its folding and restoring transcriptional activity. Its reactivation efficacy is limited to the R175H mutation, currently the third most common p53 hot spot mutation; however, other defective zinc-binding mutants such as C176, C242, C238, H179, and M237 may also be restored by ZMC-1 [109].
2.3. Restoring Downstream p53 Pathways
Sidestepping the dysfunctional mutp53 and directly remediating p53 downstream signaling is another attractive strategy. The p63/p73 belongs to pathways downstream of p53 and shares structural and functional similarity to wild-type p53, capable of tumor suppression. However, p63/p73 does not interact with wild-type p53. In contrast, some mutp53s (such as R270H) do bind to and exert dominant negative effects on p63/p73 [110]. A small molecule RETRA was shown to inhibit mutp53-p73 complex formation, thereby restoring the tumor suppressive function of native p73 in mutp53-bearing cells while sparing normal cells [111].
Activating Transcription Factor 3 (ATF3) is another druggable target that can restore p53 downstream pathway. ATF3, a stress-induced transcription factor, is known to compete with MDM2 for p53, thus preventing ubiquitination of wild-type p53 [112]. However, more recently, ATF3 was also shown to prevent mutp53 (R175H and R273H) from binding to p63 in CRC cell lines, thereby restoring the p53 downstream pathway [113]. Inhibiting Hsp90 restores ATF3 expression and confers tumor suppression [114]. Antitumor actions of Hsp90 inhibition could stem from the synergetic effects of increased mutp53 degradation and ATF3 upregulation. Patulin, a fungal toxin, can also increase ATF3 expression in colorectal cancer cell lines and induce cell apoptosis, possibly via reactive oxygen species signaling [115].
2.4. Targeting p53 Mutant Neoantigen for Immunotherapy
Other emerging strategies against mutp53s-bearing tumors, albeit ‘out of the box’ compared with the abovementioned strategies, aim to exploit the immunogenicity of mutp53s. The hypothesis is that once antibodies and/or tumor-infiltrating lymphocytes (TIL) recognize p53 neoantigens on the MHC complex, either direct or indirect immune response can be elicited to eliminate mutp53-expressing cells. Although anti-mutp53 autoantibodies can be detected in the serum of individuals with cancer, high antibody titers correlate with poor prognosis instead of improved outcome [116,117,118,119]. The lack of protective efficacy in anti-mutp53 autoantibody was attributed to improper antibody targeting to the amino and carboxyl termini of mutp53 instead of the central mutation hotspot [118]. Using a large phage library screen, Hsiue et al. [120] identified an antibody that uniquely recognizes mutp53 (R175H) but not wild-type p53. They cloned this specific antigen binding site into one end of a novel bi-specific antibody for drug targeting, and incorporated antigen specificity for T cell-CD3 complex on the other end of the antibody to elicit host lymphocytic immune response. In vivo efficacy of this bi-specific antibody was evaluated against a mutp53-bearing myeloma cell line (KMS26) implanted in mice with a reconstituted human immune system. The bi-specific antibody suppressed tumor growth but expectedly requires the presence of human T cells. This exciting finding awaits confirmation in clinical trials. Along the same design rationale, in the hope of identifying TCR specific for mutp53 neoantigen and also to expand TIL for cell-based therapy, an ongoing NCI study [121] seeks to identify tumor-infiltrating lymphocytes that recognize mutp53 neoantigens. The field of immunotherapy holds great promise as it specifically directs cytotoxicity at cancers. Since hotspot mutations change p53 structure, it is likely other hotspot mutations will exhibit unique antigens that can be targeted immunologically. Potential limitations include the high cost of drug development and testing, side effects resulting from immune activation, and the likely constraints in individuals with germline p53 mutations.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14010219
This entry is offline, you can click here to edit this entry!