Iron is an essential trace element important for many biological functions, including the essential functions of the nervous system. Understanding the mechanisms of iron homeostasis is of clinical relevance since either depletion or accumulation of intracellular iron may affect the normal function of the cell. Brain iron metabolism is a topic of growing interest since numerous researches have proved its role in neurodegeneration (e.g. Alzheimer's disease, Parkinson's disease) and other neurological disorders, especially in elderly. Maintaining of brain iron homeostasis is very specific because of the blood-brain barrier. It is a physiological barrier formed by the endothelial cells that line cerebral microvessels and has an important role in maintaining a precisely regulated brain microenvironment. Despite the years of research, the complex mechanisms for the iron influx, efflux, and regulation in the brain are not completely resolved. It would be of great importance for the future research to determine the exact cellular and molecular mechanisms related to disturbances of brain iron metabolism, especially in the context of brain aging and as potential therapeutic targets for neurodegenerative diseases.
- iron homeostasis
- brain
- iron defficiency
- iron overload
- blood-brain barrier
- neurodegeneration
- aging
1. Introduction
Iron is an essential micronutrient because of its importance in the process of erythropoiesis, oxidative metabolism, and cellular immune responses [1]. Healthy adults contain 4–5 g of iron, which is mostly (65%) found in red blood cell hemoglobin (Hb), and 30–35% is stored in the liver in the form of ferritin. Only 1–2% is in the form of iron-sulfur clusters or heme in the enzymes and multiprotein complexes [2]. Despite its essential role in the human body [3], there are no effective means of excreting iron [4]. Thus, a critical point in iron homeostasis is the regulation of the absorption of dietary iron from the duodenum. The body absorbs 1–2 mg of dietary iron a day, and this intake must be balanced with losses in the form of sloughed intestinal mucosal cells, menstruation, and other blood losses [5]. Maintaining the balance is very important because free iron is able to generate free radicals through Fenton reaction, and it is highly toxic [6][7]. Therefore, organisms have developed sophisticated pathways to import, chaperone, sequester, and export iron in order to maintain an appropriate iron balance [8]. Any disruption of iron homeostasis leads to either iron deficiency (ID) or iron overload (IO) [9].
The pathophysiological consequences of ID are impairments in cognitive development, cardiovascular diseases, endothelial disorders, and other health complications [10], especially in elderly. A chronic low-grade inflammation present in the elderly leads to less efficient absorption through hepcidin regulation and subsequent increase in ferritin concentrations. ID becomes more of a problem because of age-related changes in Hb and sex hormones, effects of medication prescribed for age-related diseases, and metabolic changes associated with inflammatory states [11].
IO leads to adverse manifestations in different tissues (brain, heart, liver, adipose, muscle, pancreas) and it is implicated in the pathogenesis of several metabolic (e.g., type 2 diabetes, non-alcoholic steatohepatitis, atherosclerosis, stroke, etc.) [12] and neurodegenerative diseases (e.g., Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), etc.) [13], which could be found more often in elderly.
Although physiological iron requirements do not differ between adult and elderly men, and post-menopausal and elderly women, there is growing evidence that iron metabolism is affected by the aging process [11]. Age-related cellular and molecular alterations in iron metabolism can lead to iron dyshomeostasis and deposition [14]. Iron deposits can contribute to the development of inflammation, abnormal protein aggregation and degeneration, especially in the central nervous system (CNS), leading to the progressive decline in cognitive processes [13], and contributing to pathophysiology of stroke [15].
An increased number of recent experimental and clinical discoveries about the sex-related differences in iron metabolism present in certain neurological disorders [16][17][18][19][20][21][22] is raising attention, and pointing to the need for future research exploring possible underlying mechanisms, which could be responsible for the sex differences in the susceptibility to these diseases.
Whereas environmental factors are involved in most of the cases of neurodegenerative diseases, it is important to take into consideration the role of nutrition in both neuroprotection and neurodegeneration. Furthermore, there is sufficient evidence regarding ID having adverse health effects to justify correcting it through diet or iron therapy. At the same time, it is important to ensure that the risk of high body iron stores is not increased as this may have detrimental effects on the brain and cause neurodegeneration and other neurological disorders [23][24]. Therefore, the aim of this review is to provide an up-to-date discussion in iron metabolism, age-related changes, along with the sex differences in iron content in serum and brain, within the healthy aging population and in neurological disorders such as MS, PD, AD, and stroke.
2. Brain Iron Metabolism
The brain is a very metabolically active organ and accounts for about 20% of the body’s total energy consumption. These high-energy needs must be supported with an adequate supply of iron [25]. Therefore, iron is the most abundant metal in the brain [14]. It has an essential role as a co-factor for many physiological processes in the CNS, including oxidative metabolism, myelination, and the biosynthesis of neurotransmitters [26]. To ensure the normal course of these processes, brain iron levels are tightly regulated [27].
The entry of iron from the blood into the brain is controlled by the blood–brain barrier (BBB) [28] and to a lesser extent by the blood–cerebrospinal fluid barrier (BCSFB) [29]. The role of the BBB is to prevent the brain from neurotoxic plasma components and pathogens [30]. At the same time, it controls chemical composition of the neuronal milieu by regulating the transport of molecules required for normal neuronal functioning [31]. The BBB is formed by a monolayer of tightly sealed microvascular endothelial cells extending along the vascular tree [32] and expressing low paracellular and transcellular permeability [33]. Those endothelial cells are surrounded by basal lamina and astrocytic perivascular end-feet, forming the neurovascular unit [34]. Tf-bound iron cannot cross the BBB directly and the mechanism of iron transcellular entry into the brain is not entirely clear [35]. According to the recent models [36][37], there are two possible iron transport pathways: transferrin-bound iron (Tf-Fe) and non-transferrin-bound iron (NTBI) [35] (Figure 1).
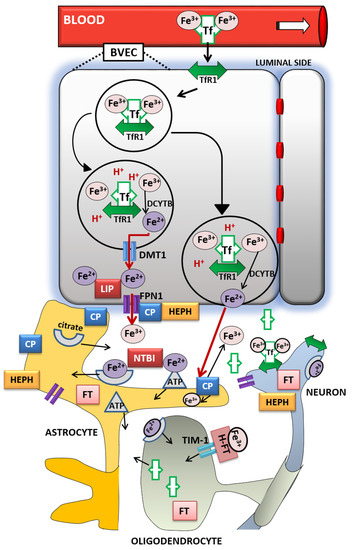
The Tf/TfR1 pathway is considered to be the major route for iron transport across the luminal membrane of the capillary endothelium [35]. According to the widely established transcytosis mechanism, Tf binds to TfR at the luminal side of the brain capillaries [38]. The complex traverses the cell in the endocytosis vesicle, where the acid environment facilitates the release of ferric iron from Tf and its reduction to ferrous iron by endosomal reductase [39], possibly duodenal cytochrome b (DCYTB) or six-transmembrane epithelial antigen of the prostate-2 (STEAP2) [40]. The next steps in this pathway are still not completely clear. One possibility is that ferrous iron is transported from the endosome to the cytosol by the DMT1 [41] and joins the intracellular labile iron pool (LIP) [42] (Figure 1). It could be further utilized for metabolic purposes by the endothelial cells, stored in endothelial cell ferritin [43] or imported into mitochondria via mitoferrins and TfR2 [44]. It could be also released into the extracellular fluid by action of export protein ferroportin (FPN1) [36], and reoxidized to Fe3+ by ferroxidases hephaestin (HEPH) and ceruloplasmin (CP) [28]. Studies have confirmed that capillary endothelium of the BBB, neurons, and astrocytes, has the ability to express FPN1 and HEPH [45][46]. The alternative mechanism that has been proposed is that the endosome containing Tf-TfR1 complex goes all the way to the abluminal side and releases iron between the endothelial cells and astrocyte end-foot processes [27]. The released ferrous iron is then oxidized to ferric iron by the ferroxidase activity of CP or HEPH expressed on the end-foot processes [41]. Oxidized iron binds to apo-Tf circulating within the brain [42] (Figure 1). The main source of Tf in the brain interstitium is its diffusion from the ventricles and a certain amount is synthesized in oligodendrocytes [47]. Because of the low concentrations of Tf in the cerebrospinal fluid (CSF), iron saturation of CSF Tf is almost 100%, while serum Tf is saturated by about 30% [27]. Consequently, under conditions of IO, CSF Tf has much lower buffering capacity [48], NTBI levels may be quite high [49], and the vulnerability of neuronal cells to iron toxicity increases [48].
Iron may also enter the brain through epithelial cells of the choroid plexus, which form the BCSFB [50]. The choroid plexus consists of fenestrated capillaries so the holo-Tf can cross them and reach the choroidal epithelium [51]. Further, the iron is released the same way as from the BBB endothelial cells by means of DMT1, FPN1 and ferroxidases [14]. When iron enters the CSF, there is no diffusional barrier between CSF and interstitial fluid. Iron binds to Tf in CSF and supplies CNS cells expressing TfR1 [52].
Different cell types in the brain acquire iron by distinct pathways. Neurons express high levels of TfR1. Therefore, Tf is the main source of iron for neurons [41], although neurons can also uptake NTBI from interstitial fluid [53]. Unlike them, oligodendrocytes and astrocytes do not express TfR1 and their main source of iron is NTBI [39]. Namely, ferrous iron in the brain interstitium can also bind to ATP or citrate released from astrocytes and it is transported to oligodendrocytes and astrocytes as NTBI [27] (Figure 1).
Oligodendrocytes acquire NTBI via the T-cell immunoglobulin and mucin domain (Tim-1). It is a ferritin receptor exclusively expressed in oligodendrocytes that binds H-ferritin [54]. Astrocytes express ferri-reductase on their plasma membranes to reduce ferric to ferrous iron and facilitate iron uptake [55] (Figure 1). Once iron enters the brain cells, the iron pool is tightly regulated. It has to provide enough iron for cellular functions and prevent the development of oxidative stress [39]. Ferritin has an important role in iron sequestration and free iron level reduction [56], whereas neuromelanin captures large amounts of iron in certain neurons for longer-term storage [57]. Namely, the pigment neuromelanin acts as a scavenger binding redox-active metal ion such as iron. The expression of ferritin varies in different cell types according to their functional requirements for iron. Neurons contain the least, and microglia contain the most amount of cytosolic ferritin [58] but in the hypoxic conditions ferritin synthesis increases in cortical neurons and decreases in glial cells [59]. Ferritin degradation by the autophagy-lysosome system [60] initiates iron release, mainly through FPN1 [61]. Since hepcidin regulates the expression of FPN1, it modulates cellular iron level as well [13]. Recent studies revealed that hepcidin can be produced by the brain endothelium [36] or systemically derived by passing the BBB [62], and it is widely distributed in the brain [63][64]. Hence, hepcidin may be involved in the regulation of iron availability and circulation in the brain [36]. Cellular iron levels are also modulated at the post-transcriptional level by binding to the iron response elements (IREs) of mRNA of (iron response proteins (IRPs) [13].
When some of these cellular and molecular mechanisms of iron regulation are disrupted, the brain iron homeostasis is disturbed as well. If there is either too much or too little iron in the brain, numerous neurologic disorders can occur [14]. Excessive brain iron accumulation is found in MS, PD and AD, amyotrophic lateral sclerosis (ALS), neurodegeneration with brain iron accumulation, and Huntington’s disease [43]. ID is associated with significant cognitive, performance and brain structural deficits [65].
3. Conclusions
Older age is associated with increased risk of ID, elevated body iron stores and increased brain iron levels [23]. Inadequate iron supply, which often accompanies aging, leads to cerebral hypoxia [66], insufficient neurotransmitter synthesis [13], impaired myelination [67], and consequently to poorer cognition, cognitive decline, and dementia [68]. On the other hand, brain iron accumulation is considered as a hallmark of aging [69] and it is associated with the progressive imbalance between antioxidant defenses and intracellular generation of ROS [70]. This increases susceptibility of aged brain to diseases and thus makes aging a major risk factor for neurodegenerative diseases development [14]. Therefore, it would be very important for the future research to determine the exact cellular and molecular mechanisms related to perturbations in iron metabolism in the aging brain to distinguish between physiological and pathological aging and find possible therapeutic targets for neurodegenerative diseases.
To counteract the ID during aging, one should certainly consider iron supplements recommended by a physician to correct the anemic state.
However, it should be noted how this supplementation may not be warranted for healthy elderly people consuming a balanced diet. In contrary, it could be detrimental for those who are homozygous or heterozygous for the HFE mutations, since recent studies showed that even moderate increases in body iron may increase the risk for body disorders including neurological ones [71], or cause irreparable damage to the brain neurons [72][73]. Because of that, older people should be careful consuming a high-iron content diet as well.
The major unknown is still the sex-related differences in iron metabolism that come with aging. Increasing experimental and clinical evidence support the idea that neurological disorders differ between women and men, suggesting the existence of different underlying mechanisms involved in their pathogenesis [16][17][18][19][20][21][22]. However, we are still far away from the actual understanding of what underlies these differences. We need a better understanding of the underlying mechanisms of how sex hormones can influence the iron metabolism and further, the development of neurological disorders. New insights into aging processes, which include the impact of sex hormones on iron metabolism as well, could enlighten the understanding of these differences during aging.
This entry is adapted from the peer-reviewed paper 10.3390/nu12092601
References
- Munoz, M.; Villar, I.; Garcia-Erce, J.A. An Update on Iron Physiology. World J. Gastroenterol. 2009, 15, 4617–4626.
- Darshan, D.; Frazer, D.M.; Anderson, G.J. Molecular Basis of Iron-Loading Disorders. Expert Rev. Mol. Med. 2010, 12, e36.
- Sheftel, A.; Stehling, O.; Lill, R. Iron-Sulfur Proteins in Health and Disease. Trends Endocrinol. Metab. 2010, 21, 302–313.
- Chen, C.; Paw, B.H. Cellular and Mitochondrial Iron Homeostasis in Vertebrates. Biochim. Biophys. Acta 2012, 1823, 1459–1467.
- Siah, C.W.; Ombiga, J.; Adams, L.A.; Trinder, D.; Olynyk, J.K. Normal Iron Metabolism and the Pathophysiology of Iron Overload Disorders. Clin. Biochem. Rev. 2006, 27, 5–16.
- Gammella, E.; Recalcati, S.; Cairo, G. Dual Role of ROS as Signal and Stress Agents: Iron Tips the Balance in Favor of Toxic Effects. Oxid. Med. Cell. Longev. 2016, 2016, 8629024.
- Zhao, Z. Iron and Oxidizing Species in Oxidative Stress and Alzheimer’s Disease. Aging Med. 2019, 2, 82–87.
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38.
- Ye, H.; Rouault, T.A. Human Iron-Sulfur Cluster Assembly, Cellular Iron Homeostasis, and Disease. Biochemistry 2010, 49, 4945–4956.
- Musallam, K.M.; Taher, A.T. Iron Deficiency Beyond Erythropoiesis: Should We Be Concerned? Curr. Med. Res. Opin. 2018, 34, 81–93.
- Van Rensburg, S.J.; Kotze, M.J.; van Toorn, R. The Conundrum of Iron in Multiple Sclerosis—Time for an Individualised Approach. Metab. Brain Dis. 2012, 27, 239–253.
- Fernandez-Real, J.M.; Manco, M. Effects of Iron Overload on Chronic Metabolic Diseases. Lancet Diabetes Endocrinol. 2014, 2, 513–526.
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The Role of Iron in Brain Ageing and Neurodegenerative Disorders. Lancet Neurol. 2014, 13, 1045–1060.
- Ashraf, A.; Clark, M.; So, P.-W. The Aging of Iron Man. Front. Aging Neurosci. 2018, 10, 65.
- Gill, D.; Monori, G.; Tzoulaki, I.; Dehghan, A. Iron Status and Risk of Stroke. A Mendelian Randomization Study. Stroke 2018, 49, 2815–2821.
- Ćurko-Cofek, B.; Grubić Kezele, T.; Marinić, J.; Tota, M.; Starčević Čizmarević, N.; Milin, Č.; Ristić, S.; Radošević-Stašić, B.; Barac-Latas, V. Chronic Iron Overload Induces Gender-Dependent Changes in Iron Homeostasis, Lipid Peroxidation and Clinical Course of Experimental Autoimmune Encephalomyelitis. Neurotoxicology 2016, 27, 1–12.
- Persson, N.; Wu, J.; Zhang, Q.; Liu, T.; Shen, J.; Bao, R.; Ni, M.; Liu, T.; Wang, Y.; Spincemaille, P. Age and Sex Related Differences in Subcortical Brain Iron Concentrations Among Healthy Adults. Neuroimage 2015, 122, 385–398.
- Koellhoffer, E.C.; McCullough, L.D. The Effects of Estrogen in Ischemic Stroke. Transl. Stroke Res. 2013, 4, 390–401.
- Zeller, T.; Schnabel, R.B.; Appelbaum, S.; Ojeda, F.; Berisha, F.; Schulte-Steinberg, B.; Brueckmann, B.E.; Kuulasmaa, K.; Jousilahti, P.; Blankenberg, S.; et al. Low Testosterone Levels Are Predictive for Incident Atrial Fibrillation and Ischaemic Stroke in Men, but Protective in Women—Results from the FINRISK Study. Eur. J. Prev. Cardiol. 2018, 25, 1133–1139.
- Khalifa, A.R.M.; Abdel-Rahman, E.A.; Mahmoud, A.M.; Ali, M.H.; Noureldin, M.; Saber, S.H.; Mohsen, M.; Ali, S.S. Sex-Specific Differences in Mitochondria Biogenesis, Morphology, Respiratory Function, and ROS Homeostasis in Young Mouse Heart and Brain. Physiol. Rep. 2017, 5, e13125.
- Jurado-Coronel, J.C.; Cabezas, R.; Rodríguez, M.F.A.; Echeverria, V.; García-Segura, L.M.; Barreto, G.E. Sex Differences in Parkinson’s Disease: Features on Clinical Symptoms, Treatment Outcome, Sexual Hormones and Genetics. Front. Neuroendocrinol. 2018, 50, 18–30.
- Bartzokis, G.; Lu, P.H.; Tingus, K.; Peters, D.G.; Amar, C.P.; Tishler, T.A.; Finn, J.P.; Villablanca, P.; Altshuler, L.L.; Mintz, J. Gender and Iron Genes may Modify Associations Between Brain Iron and Memory in Healthy Aging. Neuropsychopharmacology 2011, 36, 1375–1384.
- Fairweather-Tait, S.J.; Wawer, A.A.; Gillings, R.; Jennings, A.; Myint, P.K. Iron Status in the Elderly. Mech. Ageing Dev. 2014, 136, 22–28.
- Belaidi, A.A.; Bush, A.I. Iron Neurochemistry in Alzheimer’s Disease and Parkinson’s Disease: Targets for Therapeutics. J. Neurochem. 2016, 139, 179–197.
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901.
- Salvador, G.A. Iron in Neuronal Function and Dysfunction. Biofactors 2010, 36, 103–110.
- Moos, T.; Nielsen, T.R.; Skjørringe, T.; Morgan, E.H. Iron Trafficking Inside the Brain. J. Neurochem. 2007, 103, 1730–1740.
- McCarthy, R.C.; Kosman, D.J. Iron Transport Across the Blood-Brain Barrier: Development, Neurovascular Regulation and Cerebral Amyloid Angiopathy. Cell. Mol. Life Sci. 2015, 72, 709–727.
- Moos, T.; Morgan, E.H. Transferrin and Transferrin Receptor Function in Brain Barrier Systems. Cell. Mol. Neurobiol. 2000, 20, 77–95.
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s Disease: A Matter of Blood-Brain Barrier Dysfunction? J. Exp. Med. 2017, 214, 3151–3169.
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078.
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-Brain Barrier: From Physiology to Disease and Back. Physiol. Rev. 2019, 99, 21–78.
- Zlokovic, B.V. Neurovascular Pathways to Neurodegeneration in Alzheimer’s Disease and Other Disorders. Nat. Rev. Neurosci. 2011, 12, 723–738.
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey Through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42.
- Ke, Y.; Qian, Z.M. Brain Iron Metabolism: Neurobiology and Neurochemistry. Prog. Neurobiol. 2007, 83, 149–173.
- Simpson, I.A.; Ponnuru, P.; Klinger, M.E.; Myers, R.L.; Devraj, K.; Coe, C.L.; Lubach, G.R.; Carruthers, A.; Connor, J.R. A Novel Model for Brain Iron Uptake: Introducing the Concept of Regulation. J. Cereb. Blood Flow Metab. 2015, 35, 48–57.
- Khan, A.I.; Liu, J.; Dutta, P. Iron Transport Kinetics Through Blood-Brain Barrier Endothelial Cells. Biochim. Biophys. Acta 2018, 1862, 1168–1179.
- Duck, K.A.; Connor, J.R. Iron Uptake and Transport Across Physiological Barriers. Biometals 2016, 29, 573–591.
- Rouault, T.A.; Cooperman, S. Brain Iron Metabolism. Semin. Pediatr. Neurol. 2006, 13, 142–148.
- De Domenico, I.; McVey Ward, D.; Kaplan, J. Regulation of Iron Acquisition and Storage: Consequences for Iron-Linked Disorders. Nat. Rev. Mol. Cell Biol. 2008, 9, 72–81.
- Benarroch, E.E. Brain Iron Homeostasis and Neurodegenerative Disease. Neurology 2009, 72, 1436–1440.
- Burkhart, A.; Skjørringe, T.; Johnsen, K.B.; Siupka, P.; Thomsen, L.B.; Nielsen, M.S.; Thomsen, L.L.; Moos, T. Expression of Iron-Related Proteins at the Neurovascular Unit Supports Reduction and Reoxidation of Iron for Transport Through the Blood-Brain Barrier. Mol. Neurobiol. 2016, 53, 7237–7253.
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The Role of Iron and Copper Molecules in the Neuronal Vulnerability of Locus Coeruleus and Substantia Nigra during Aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848.
- Horowitz, M.P.; Greenamyre, J.T. Mitochondrial Iron Metabolism and Its Role in Neurodegeneration. J. Alzheimers Dis. 2010, 20, S551–S568.
- Wu, L.J.; Leenders, A.G.; Cooperman, S.; Meyron-Holtz, E.; Smith, S.; Land, W.; Tsai, R.Y.; Berger, U.V.; Sheng, Z.H.; Rouault, T.A. Expression of the Iron Transporter Ferroportin in Synaptic Vesicles and the Blood–Brain Barrier. Brain Res. 2004, 1001, 108–117.
- Qian, Z.M.; Chang, Y.Z.; Du, J.R.; Ho, K.P.; Zhu, L.; Xu, Y.J.; Li, L.Z.; Wang, C.Y.; Wang, Q.; Ge, X.H.; et al. Development and Iron-Dependent Expression of Hephaestin in Different Brain Regions of Rats. J. Cell. Biochem. 2007, 102, 1225–1233.
- De Arriba Zerpa, G.A.; Saleh, M.C.; Fernandez, P.M.; Guillou, F.; Espinosa de los Monteros, A.; De Vellis, J.; Zakin, M.M.; Baron, B. Alternative Splicing Prevents Transferrin Secretion During Differentiation of a Human Oligodendrocyte Cell Line. J. Neurosci. Res. 2000, 61, 388–395.
- Singh, N.; Haldar, S.; Tripathi, A.K.; Horback, K.; Wong, J.; Sharma, D.; Beserra, A.; Suda, S.; Anbalagan, C.; Dev, S.; et al. Brain Iron Homeostasis: From Molecular Mechanisms to Clinical Significance and Therapeutic Opportunities. Antioxid. Redox Signal. 2014, 20, 1324–1363.
- Nnah, I.C.; Wessling-Resnick, M. Brain Iron Homeostasis: A Focus on Microglial Iron. Pharmaceuticals 2018, 11, 129.
- Rouault, T.A.; Zhang, D.L.; Jeong, S.Y. Brain Iron Homeostasis, the Choroid Plexus, and Localization of Iron Transport Proteins. Metab. Brain Dis. 2009, 24, 673–684.
- Brown, P.D.; Davies, S.L.; Speake, T.; Millar, I.D. Molecular Mechanisms of Cerebrospinal Fluid Production. Neuroscience 2004, 129, 957–970.
- Leitner, D.F.; Connor, J.R. Functional Roles of Transferrin in the Brain. Biochim. Biophys. Acta 2012, 1820, 393–402.
- Burdo, J.R.; Menzies, S.L.; Simpson, I.A.; Garrick, L.M.; Garrick, M.D.; Dolan, K.G.; Haile, D.J.; Beard, J.L.; Connor, J.R. Distribution of Divalent Metal Transporter 1 and Metal Transport Protein 1 in the Normal and Belgrade Rat. J. Neurosci. Res. 2001, 66, 1198–1207.
- Han, J.; Seaman, W.E.; Di, X.; Wang, W.; Willingham, M.; Torti, F.M.; Torti, S.V. Iron Uptake Mediated by Binding of H-ferritin to the Tim-2 Receptor in Mouse Cells. PLoS ONE 2011, 6, e23800.
- Bishop, G.M.; Scheiber, I.F.; Dringen, R.; Robinson, S.R. Synergistic Accumulation of Iron and Zinc by Cultured Astrocytes. J. Neural Transm. 2010, 117, 809–817.
- Vidal, R.; Miravalle, L.; Gao, X.; Barbeito, A.G.; Baraibar, M.A.; Hekmatyar, S.K.; Widel, M.; Bansal, N.; Delisle, M.B.; Ghetti, B. Expression of a Mutant Form of the Ferritin Light Chain Gene Induces Neurodegeneration and Iron Overload in Transgenic Mice. J. Neurosci. 2008, 28, 60–67.
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of Iron, Dopamine and Neuromelanin Pathways in Brain Aging and Parkinson’s Disease. Prog. Neurobiol. 2017, 155, 96–119.
- MacKenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular Iron Transport and Storage: From Molecular Mechanisms to Health Implications. Antioxid. Redox Signal. 2008, 10, 997–1030.
- Irace, C.; Scorziello, A.; Maffettone, C.; Pignataro, G.; Matrone, C.; Adornetto, A.; Santamaria, R.; Annunziato, L.; Colonna, A. Divergent Modulation of Iron Regulatory Proteins and Ferritin Biosynthesis by Hypoxia/Reoxygenation in Neurons and Glial Cells. J. Neurochem. 2005, 95, 1321–1331.
- Asano, T.; Komatsu, M.; Yamaguchi, Y.; Ishikawa-Iwai, F.; Mizushima, N.; Iwai, K. Distinct Mechanisms of Ferritin Delivery to Lysosomes in Iron-Depleted and Iron-Replete Cells. Mol. Cell. Biol. 2011, 31, 2040–2052.
- Biasiotto, G.; Di Lorenzo, D.; Archetti, S.; Zanella, I. Iron and Neurodegeneration: Is Ferritinophagy the Link? Mol. Neurobiol. 2016, 53, 5542–5574.
- Vela, D. The Dual Role of Hepcidin in Brain Iron Load and Inflammation. Front. Neurosci. 2018, 12, 740.
- Zechel, S.; Huber-Wittmer, K.; Von Bohlen und Halbach, O. Distribution of the Iron-Regulating Protein Hepcidin in the Murine Central Nervous System. J. Neurol. Res. 2006, 84, 790–800.
- Wang, Q.; Du, F.; Qian, Z.M.; Ge, X.H.; Zhu, L.; Yung, W.H.; Yang, L.; Ke, Y. Lipopolysaccharide Induces a Significant Increase in Expression of Iron Regulatory Hormone Hepcidin in the Cortex and Substantia Nigra in Rat Brain. Endocrinology 2008, 149, 3920–3925.
- Beard, J.L.; Connor, J.R. Iron Status and Neural Functioning. Annu. Rev. Nutr. 2003, 23, 41–58.
- Petranovic, D.; Batinac, T.; Petranovic, D.; Ruzic, A.; Ruzic, T. Iron Deficiency Anaemia Influences Cognitive Functions. Med. Hypotheses 2008, 70, 70–72.
- Bourre, J.M. Effects of Nutrients (in Food) on the Structure and Function of the Nervous System: Update on Dietary Requirements for Brain. Part 1: Micronutrients. J. Nutr. Health Aging 2006, 10, 377–385.
- Andro, M.; Le Squere, P.; Estivin, S.; Gentric, A. Anaemia and Cognitive Performances in the Elderly: A Systematic Review. Eur. J. Neurol. 2013, 20, 1234–1240.
- Hosking, D.E.; Ayton, S.; Beckett, N.; Booth, A.; Peters, R. More Evidence Is Needed. Iron, Incident Cognitive Decline and Dementia: A Systematic Review. Ther. Adv. Chronic. Dis. 2018, 9, 241–256.
- Poon, H.F.; Calabrese, V.; Calvani, M.; Butterfield, D.A. Proteomics Analyses of Specific Protein Oxidation and Protein Expression in Aged Rat Brain and its Modulation by L-Acetylcarnitine: Insights into the Mechanisms of Action of this Proposed Therapeutic Agent for CNS Disorders Associated with Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 381–394.
- Garry, P.J.; Hunt, W.C.; Baumgartner, R.N. Effects of Iron Intake on Iron Stores in Elderly Men and Women: Longitudinal and Cross-Sectional Results. J. Am. Coll. Nutr. 2000, 19, 262–269.
- Ferreira, A.; Neves, P.; Gozzelino, R. Multilevel Impacts of Iron in the Brain: The Cross Talk between Neurophysiological Mechanisms, Cognition, and Social Behavior. Pharmaceuticals 2019, 12, 126.
- Rutten, B.P.F.; Schmitz, C.; Gerlach, O.H.H.; Oyen, H.M.; de Mesquita, E.B.; Steinbusch, H.W.M.; Korr, H. The Aging Brain: Accumulation of DNA Damage or Neuron Loss? Neurobiol. Aging 2007, 28, 91–98.