Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
Immune therapy is designed to stimulate tumoricidal effects in a variety of solid tumors including breast carcinomas. However, the emergence of resistant clones leads to treatment failure. Understanding the molecular, cellular, and microenvironmental aberrations is crucial to uncovering underlying mechanisms and developing advanced strategies for preventing or combating these resistant malignancies.
- antigen presentation and recognition
- breast cancer
- immune evasion
1. Introduction
Female breast cancer (BC) is the most diagnosed malignancy, with approximately 2.3 million new cases (11.7% of all cancer incidences) worldwide in 2020. That number is projected to increase to over 3 million by 2040, according to the International Agency for Research on Cancer (GLOBOCAN) estimates [1].
Based on the estrogen receptor (ER), progesterone receptor (PR), human epidermal growth factor receptor 2 (HER2), as well as a BC proliferation index Ki67, our understanding of BC biology reveals 4 intrinsic molecular subtypes. They include luminal A (resembling the histological phenotype: ER+, PR+, HER2−, Ki67Low), luminal B (ER+, PR+, HER+/−, Ki67High), HER2-enriched (ER−, PR−, HER2+), and basal-like subtype (ER−, PR−, HER2−) which largely resembles triple-negative BC (TNBC) and comprises approximately 15% of all BC cases [2]. The ER is found expressed in two luminal subtypes and can distinguish luminal from non-luminal malignancies [3]. Luminal A and B subtypes are characterized by their prominent activation of luminal/hormone-regulated pathways as well as proliferation/cell cycle regulators [4]. Luminal A subtype has a higher expression of luminal-related genes or proteins such as FOXA1 and lower expression of Ki67 than Luminal B [3][5]. The HER2-enriched subtype is distinguished by the high expression of HER2-related and proliferation-associated regulators such as ERBB2/HER2, insignificant expression of luminal-related genes, and negligible expression of genes related to the basal layer of the skin (e.g., keratin 5) [6]. The Basal-like subtype is characterized by high expression of Ki67 as well as keratins 5, 14, and 17, a low to undetectable expression of HER2-related genes, and unnoticeable expression of luminal-related genes [6].
The molecular subtyping helps determine the most appropriate first-line therapy. ER+ tumors are targeted with endocrine therapeutic agents such as tamoxifene, aromatase inhibitors, and abemaciclib; HER2 over-expressing tumors are generally treated with HER2-blocking antibodies such as trastuzumab and pertuzumab, whereas TNBC is treated with standard cytotoxic therapies and radiotherapy [7]. However, given TNBC is frequently resistant to chemotherapy and radiotherapy, one promising treatment regimen remaining is immuno-oncology therapeutics. Considering the higher mutational burden, TNBC is known to be the most immunogenic subtype. They are frequently associated with tumor-infiltrating lymphocytes (TIL) indicative of a favorable prognosis [8].
All human nucleated cells process their intracellular proteins through the proteasome system and then present the degraded peptide fragments (the epitopes) on the major histocompatibility complex (MHC)-I. This immune complex is then scrutinized by surveillance lymphocytes. Any circulating activated T lymphocytes encountering the non-self or abnormal moieties within the peptide-MHC-I complex will either directly eliminate the target cell or produce inflammatory cytokines. After recognizing a malignant antigen, a cluster of differentiated (CD)4+ T helper (Th) cells can secrete pro-inflammatory cytokines to recruit additional immune cells and mount an immune response, whereas CD8+ cytotoxic T lymphocytes (CTL) can directly destroy tumor cells by secretion of cytotoxic molecules such as granzymes leading to apoptosis [9]. A successful antitumor immune response requires the following key phases: (1) capture of tumor antigens (or epitopes) by antigen-presenting cells followed by presentation them to lymphocytes; (2) activation and expansion of CD4+ and/or CD8+ lymphocytes; (3) secretion of inflammatory cytokines by CD4+ lymphocytes and destruction of tumor cells by CD8+ lymphocytes with involvement of dendritic cells (DCs), natural killer (NK) cells, and macrophages [10][11]. However, the tumoricidal immunity can be muted by aggressive or metastasized cancer cells leading to a detrimental phase known as “immune editing” and “immune tolerance” [12].
2. Initial Anti-Tumor Immunity Defeats Breast Neoplasm
Breast tumors are not solely masses of excessively proliferating cells. Instead, they are intermingled with a repertoire of resident and recruited non-cancerous cells such as fibroblasts, endothelial and immune cells. These cells, along with their secreted soluble factors as well as the insoluble extracellular matrix proteins collectively constitute the tumor microenvironment (TME) [13].
The initial combined innate and adaptive immune responses are designed to eradicate tumor growth. This early phase comprises acute inflammatory reactions in response to tumor cell recognition, the secretion of proinflammatory cytokines, and the destruction of malignant cells by innate immune cells such as NK cells, DCs, and macrophages. Upon maturation, antigen-presenting cells (APCs; predominantly DCs and macrophages) migrate to nearby lymph nodes (LN) where they recognize, internalize, digest, and then present tumor antigens at the cell surface with MHC-I or MHC-II [14][15]. Next, the epitope-bound APCs activate tumor-specific CD4+ Th cells and CD8+ CTL that migrate to the tumor site and assist in killing. The activated T cells differentiate, proliferate, and prime for effector functions that inspect cells expressing tumor-associated antigens. After the initial immune surveillance, the tumor cells are either completely eradicated or a few immune-evading clones merge [16][17].
3. Breast Cancer Reprograms Tumoricidal Immunity
Leukocytes from the innate and adaptive immune systems participate not only in initial tumoral rejection but also during tumor growth progression and metastatic spread [12][18]. In the early phase of tumor evolution, host immune factors, in particular cells of the innate immune system, play a key role in the elimination of tumor cells [19]. However, in the equilibrium phase, tumor cells are maintained in a dormant state [20]. This progresses to an immune escape when the tumor variants emerge, blunting immune recognition and establishing an immunosuppressive TME [21]. Overall, these actions are consistent with the repertoire of immunoediting whereby aggressive tumors established distinct mechanisms to evade immune surveillance, establish immune tolerance, and promote their proliferation.
3.1. Aberrant Presentation of Tumor-Associated Antigens (TAA)
A crucial process for T cell recognition of tumors is MHC-binding of TAA peptides with a presentation on the surface of tumor cells or APCs. In TNBC, high levels of the MHC-II antigen presentation pathway were found to be correlated with favorable progression-free survival, reduced rate of relapse, and abundant infiltration of CD8+ CTL [22]. Conversely, defects in the antigen processing and presenting machinery (APM) diminish tumor cell recognition and killing by CD8+ CTL. Awareness of the functionality of APM is important when administering T cell-based immunotherapy protocols [23]. Furthermore, not all mutated proteins are recognized equally by T cells. For T cell recognition, neoantigens should be processed in short peptides of 9–15 amino acids. As they vary in length, only a fraction are eligible to trigger immune recognition [24].
The APM is suppressed by the expression of myelin and lymphocyte protein 2 (MAL2). Initially noted in hepatoma, MAL2 encodes a transmembrane protein associated with protein endocytosis, mainly by aiding the delivery of membrane-bound proteins and exogenous cargos from the basolateral to the apical surface [25]. Using human and mouse BC cell lines, Fang et al. demonstrated that by interacting with partner effectors, RAB7 and MHC-I molecules, MAL2 augments the endocytosis of MHC-I molecules to the late-stage endosome for degradation, downregulates CD8+ T cell cytotoxicity, and thus weakens immune recognition [26] (Figure 1A). High expression of MAL2 in BC decreases the stability and the level of the antigen-loaded MHC-I on the cell membrane, leading to poor antigen presentation as well as diminished cytotoxicity response from CD8+ CTL [26]. MAL2high tumors escape recognition by CD8+ CTL cells, thereby greatly increasing immune tolerance and worsening disease prognosis [26]. In another parallel study, expression of the transport-associated proteins (TAP1/TAP2), required for proper antigen loading, is concordantly down-regulated in high-grade BC [27]. Apart from downregulating gene expression, mutations affecting antigen presentation also provide another independent mechanism of immune escape. Mutations in β2-microglobulin (B2M), a component of MHC-I, are shown to render immune suppressive effects and thereby become a potential target for therapy [28][29].
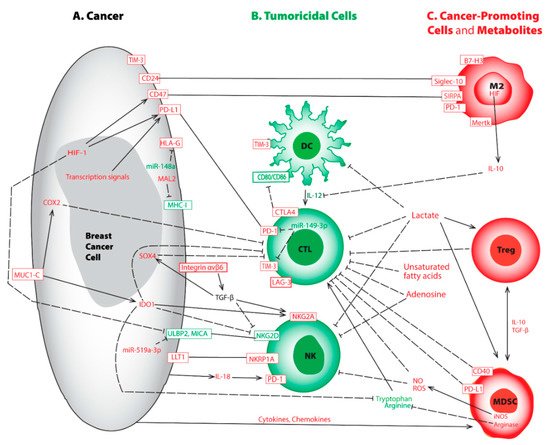
Figure 1. Representative aberrations in breast cancer cell (A), tumoricidal cells (B), cancer-promoting cells and metabolites (C) lead to immune evasion. This crosstalk map indicates the tumoricidal cells and factors (in green) and the pro-tumorigenic counterparts and factors (in red). Rectangular boxes represent cell surface molecules; plain lines indicate binding; solid arrows represent activation; and dashed lines show inhibition between modulators. Abbreviations used include cluster of differentiated (CD), CTL-associated protein 4 (CTLA-4), cyclooxygenase-2 (COX2), cytotoxic T lymphocyte (CTL), dendritic cell (DC), human leukocyte antigen G (HLA-G), hypoxia-inducible factor (HIF), indoleamine 2,3-dioxygenase (IDO), inducible nitric oxide synthase (iNOS), interleukin (IL), lymphocyte activation gene-3 (LAG-3), lectin-like transcript-1 (LLT1), M2 macrophage (M2), myelin and lymphocyte protein 2 (MAL2), major histocompatibility complex class I (MHC-I), myeloid-derived suppressor cells (MDSC), macrophage c-mer tyrosine kinase (Mertk), major histocompatibility complex (MHC), MHC-I related chain A (MICA), mucin 1 C-terminal (MUC1-C), natural killer cell (NK), nitric oxide (NO), reactive oxygen species (ROS), regulatory T cell (Treg), programmed death receptor-1 (PD-1), programmed death-ligand 1 (PD-L1), sialic acid binding Ig like lectin-10 (Siglec-10), signal regulatory protein α (SIRPA), T-cell immunoglobulin and mucin domain-containing molecule 3 (TIM-3), transforming growth factor β (TGF-β), and UL16 binding protein 2 (ULBP2). It is worth mentioning the sizes of various cells may be disproportional to their actual dimensions.
On the other hand, studies on high-grade BC demonstrate up-regulations of non-classical human leukocyte antigen (HLA)-E, HLA-F, and HLA-G are important for promoting immune escape [30][31][32]. In BC, elevated expression of HLA-G not only renders resistance towards neoadjuvant chemotherapy (NAC) [33], but also correlates with poor prognosis [33][34][35][36]. Another independent investigation on ER+ BC demonstrates that estrogen signaling silences a microRNA known as miR-148a that further elevates HLA-G expression and promotes immune evasion [37] (Figure 1A; Table 1). HLA-G conveys suppressive effects on adaptive and innate immunity by interacting with immune cell inhibitory receptors such as leukocyte immunoglobulin-like receptors B1 and B2 (LILRB1 and LILRB2) or killer cell immunoglobulin-like receptor 2DL4 (KIR2DL4) [38].
3.2. Dysfunctional CD8+ Tumor-Infiltrating Lymphocytes (TIL)
NAC-induced tumor cell death results in increased TIL largely due to the release of processed antigens from dead tumor cells followed by APC uptake and presentation [39]. Among lymphocytes, CTL is associated with a favorable prognosis, and high frequencies (>60%) of TIL are predictive for therapy response to NAC [40]. Recent studies among BC subtypes demonstrate TNBC often harbors the highest numbers of TIL and they are associated with neoantigens. However, the intra-tumoral CD8+ CTL may present at the exhausted phase due to prolonged exposure to immune-suppressive molecules in the TME [17][41][42]. Exhausted T cells neither produce antitumor cytokines nor execute their tumoricidal functionality [43][44]. Bagati et al. demonstrate, via the integrin αvβ6–TGF-β–SOX4 signaling pathway, SOX4 expression is upregulated by the integrin αvβ6 receptor on the surface of TNBC tumor cells thereby changing transforming growth factor (TGF)-β from a latent precursor to an active form [45] (Figure 1A; Table 1). High expression of SOX4 not only blocks tumoricidal function normally associated with CTL but also correlates with a poor prognosis [45]. Antibody-mediated blockade of integrin αvβ6 attenuates SOX4 expression and restores T cell-mediated cytotoxicity [45]. Likewise, SOX4 pathway inhibition prevents the emergence of MHC-ILow tumor cells that are also refractory to CD8+ CTL [45].
In addition, mounting evidence indicates interferon (IFN)-γ signaling, when chronically activated in tumors, conveys an immune-suppressive TME [46][47]. Notably, the oncogenic protein mucin 1 C-terminal (MUC1-C) is aberrantly produced by TNBC, and its expression leads to both depleted and dysfunctional CTL [48][49]. Molecular studies reveal MUC1-C induces the IFN-γ→JAK1→STAT1→IRF1 signaling cascade as well as the downstream indoleamine 2,3-dioxygenase (IDO)1 and cyclooxygenase (COX)2 effectors, thereby synergistically inhibiting CD8+ T cells in TNBC [42] (Figure 1A; Table 1). Upregulation of IDO1 in TNBCs lowers the levels of tryptophan, an amino acid essential for proper T-cell proliferation and immune function in the TME [50] (Figure 1A). Similarly, elevated COX2 expression in TNBCs increases the production of prostaglandin (PG) E2 leading to T-cell dysfunction [51][52]. Targeting MUC1-C could be of clinical importance by disrupting this immunosuppression niche [42].
3.3. Aberrant Immune Checkpoint Modulators
Immune responses are controlled by a plethora of checkpoint regulators that act as “security brakes”, to terminate immune reactions when an infection is resolved, to promote self-tolerance, and to protect against autoimmunity. Tumors exploit such immune checkpoint molecules, attempting to dampen antitumor responses, favor immune tolerance, and escape recognition and destruction [53]. Blocking the activity of one or several of these immune checkpoint molecules is shown to rescue otherwise exhausted antitumor T cells and, most importantly, to improve clinical outcomes as well as survival benefits in cancer patients [54].
3.3.1. CTLA-4
At the anti-tumor immune surveillance stage, T lymphocyte activation requires recognition of peptide-loaded MHC-I by the T-cell receptor (TCR) followed by binding between CD28 on T-lymphocytes and the counterpart ligands CD80/CD86 on APCs. However, in BC, neoantigen recognition may result in the emergence of an inhibitory receptor known as CTL-associated protein 4 (CTLA-4), which is translocated to the cell surface. CTLA-4 is a homolog of CD28, and it harbors a strong affinity for binding CD80/CD86. This interaction not only hijacks the activation signal normally executed by CD28 binding but also unleashes a contrary signal that abrogates CTL function [55] (Figure 1B, Table 1). By triggering a negative feedback loop and weakening the immune surveillance effect, CTLA-4 is widely recognized as a crucial regulator of T cell self-tolerance and immune evasion leading to poor prognosis [56][57].
3.3.2. PD-1 and PD-L1
Programmed death receptor-1 (PD-1) is an inhibitory transmembrane protein expressed on T cells, B cells, macrophages, and NK cells. The interaction between PD-1 and its ligand programmed death-ligand 1 (PD-L1) on tumor cells, ignites a “stop eating me” signal that directly contributes to immune evasion via promotion of peripheral T effector (Teff) cell exhaustion and conversion to immunosuppressive T regulatory (Treg) cells, thereby hindering tumor destruction [53][58] (Figure 1A,B; Table 1). PD-L1 expression ranges from 20% to 50% in all BC subtypes and is higher in TNBC patients as compared to non-TNBC [59][60][61]. High levels of PD-L1 are associated with poor overall survival (OS) [62], and elevated PD-L1 expression is involved in immune evasion and poor prognosis in TNBC [63].
Importantly, responses to checkpoint immunotherapy could be modulated by expression levels of PD-L1 on tumor cells. For instance, PD-L1 can be palmitoylated in its cytoplasmic domain, and this lipid modification sustains PD-L1 stability by preventing ubiquitination and subsequent degradation. Inhibition of palmitoylation shortens the lifespan of PD-L1 and enhances T cell-mediated tumoricidal activity [64] (Table 1). This finding is substantiated by Nouri et al. who elucidated the Hippo pathway effector, yes-associated protein, and transcriptional co-activator with PDZ-binding motif (YAP/TAZ) is critical in mediating anaplastic lymphoma kinase (ALK)-induced up-regulation of PD-L1 in multiple cancer cell lines. Knocking down YAP/TAZ impedes ALK-mediated immune evasion due to lowered PD-L1 expression [65]. Moreover, in a human BC stem cell model, elevated PD-L1 correlated with promoter CpG de-methylation and aberrant posttranslational histone modifications comprising lowered occupancy of repressive histones in the PD-L1 promoter region and overexpression of histone acetylation enzymes [66]. Further studies on the stem cell-enriched fraction of TNBC reveal that elevated PD-L1 expression can be induced by the Wingless/int1 (WNT) signaling pathway [67]. In addition, BRD4, a member of the bromodomain and extra-terminal domain, can transcriptionally up-regulate PD-L1 expression by binding to its promoter [68][69].
It is worth noting that in TNBC, the signal transducer and activator of transcription (STAT)-3 and its homolog STAT1 are also involved in regulating PD-L1 expression. Mechanistic studies show that phosphorylated STAT1 binds phosphorylated STAT3 in the cytoplasm, and the complex translocate into the nucleus where this heterodimer binds the PD-L1 promoter and activates its transcription [70] (Table 1). Initially, syntenin1 was shown to induce T cell exhaustion in vivo. Further studies linked STAT3 with syntenin1 and demonstrated the syntenin1-driven, STAT3-dependent signaling cascade, can upregulate PD-L1 in a TNBC model [71]. Taken together, it is rationalized that targeting syntenin1, in conjunction with inhibiting STAT3, could become a potential strategy for restoring CTL activity and thereby improving the prognostic outcomes of patients with TNBC. Additional studies demonstrate the expression of PD-L1 can be elevated by Crk in TNBC [72], by neuromedin U (NmU) in HER2+ BC [73], or by exposure to radiation [74].
Given the PD-1/PD-L1 association inactivates T cells and attenuates tumoricidal effects, disrupting PD-1/PD-L1 binding may evolve into a promising therapeutic regimen. As of March 2019, the United States Food and Drug Administration (FDA) had approved seven immune blockade therapies for treating a variety of cancers including BC. The antibodies primarily target two major immune checkpoint pathways, PD-1, and PD-L1, as well as CTLA [75][76]. Accordingly, the 1st line immune checkpoint blockade therapy in combination with chemotherapy was administered for treating TNBC patients who express PD-L1 [77]. For patients who respond poorly or develop secondary resistance to immunotherapy, treatments will combine with additional epigenetic agents [78][79], or with chemotherapeutic agents [80]. Current investigations are underway to determine the benefits of combining immunotherapy with radiation [81][82][83].
3.3.3. LAG-3
Lymphocyte activation gene-3 (LAG-3) is a member of the immunoglobulin superfamily that was first identified in 1990 [84]. LAG-3 is expressed on activated CD4+ and CD8+ T cells and on a subset of NK cells. It is structurally similar to CD4 and binds MHC-II with a higher affinity than CD4 [85][86]. Although LAG-3’s mechanism of action remains to be elucidated, it is certain that increased expression of LAG-3 can be triggered in chronically activated T cells [87][88]. LAG-3 exerts a remarkable synergy with PD-1 to transduce the inhibitory impact on activated CD8+ T cells (Figure 1B). This leads to immune escape [89][90][91] through the interaction of LAG-3’s ligands available in the TME [92][93]. LAG-3 and PD-1 have been shown to be co-expressed on TIL, and blockade of both regulators had synergistic effects on restoration of the anti-tumor CD8+ T cell response [90][91] (Figure 1B; Table 1).
3.3.4. TIM-3
T-cell immunoglobulin and mucin domain-containing molecule 3 (TIM-3), initially identified based upon its expression on CD4+ Th1 cells and CTL [94], is another immune checkpoint modulator that contributes to immune suppression in BC [95]. TIM-3 hampers proliferation attenuates the production of effective cytokines and augments apoptosis of effector T cells, through interaction with its ligands galectin-9, high mobility group protein B1 (HMGB1), carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM-1), and phosphatidylserine [95][96][97]. TIM-3 is expressed on a variety of immune cells including T lymphocytes, DCs, and BC cells [98] (Figure 1A,B).
Upon studying DCs, blockade of TIM-3 by introducing anti-(α)TIM-3 antibodies improves response to paclitaxel chemotherapy in murine models of luminal B and TNBC diseases [99]. Furthermore, through prolonged exposure to IL-12, combined efficacy not only boosts the effector function of intertumoral CTL but also elevates granzyme B expression with minimal cytotoxicity [99] (Table 1). Another independent study shows TIM-3 inhibits the production of the chemokine CXCL9/10 by DCs thereby limiting antitumor immunity in mammary carcinomas [100]. TIM-3 blockade, similarly, enhances response to paclitaxel, augments uptake of extracellular DNA by DCs through an endocytic process, and renders re-activation of the cytoplasmic DNA-binding by HMGB1 as well as sensing by the cyclic GMP-AMP synthase (cGAS) and stimulator of IFN genes (STING) pathway in DCs [100]. Together, upon TIM-3 blockade, elevated chemokines released from DCs can strengthen T cell effector function and response to chemotherapeutic treatment [99][100]. The immune surveillance function of DCs within tumors is emerging as a critical determinant of an effective T cell response [101], and TIM-3 inhibition depicts not only chemotherapeutic susceptibility but also promising efficacy in cancer immunotherapy.
On the other hand, TIM-3 overexpression in BC cells promotes cell proliferation, migration, invasion, and enhances chemoresistance to paclitaxel through the overly activated NF-κB/STAT3 pathway [102]. STAT3 signaling displays a plethora of roles in immune cells and promotes the immunosuppressive function in the TME [103]. TIM-3 overexpression in BC cells activates the STAT3 signal pathway that promotes crosstalk between cancer and immune cells [104]. TIM-3 also destroys tight junctions, which further accelerates cancer progression [102]. Conversely, downregulation of TIM-3 in BC cells inhibits their proliferation, migration, invasion, and promotes apoptosis [105].
One of the microRNAs, miR-149-3p, has been reported to bind 3′UTRs of mRNAs encoding PD-1, TIM-3, and other immune checkpoints [106]. Treatment of CTL with miR-149-3p mimic rescues T-cell exhaustion, downregulates PD-1 and TIM-3, and thereby promotes the killing of 4T1 mouse breast tumor cells [106] (Table 1). Moreover, TIM-3 has recently emerged as a promising target for cancer immunotherapy, because it is a non-redundant regulator that differs from other better-characterized checkpoints. Several prospective studies and clinical trials have been launched in solid tumors [107]. αTIM-3 partially reverses this exhausted phenotype, results in improved expression of IFN-γ, and suppresses tumor growth in multiple preclinical models [108]. αTIM-3 antibodies have revealed successful efficacious treatment synergy when combined with αPD-1 [109] or when it is used subsequently in αPD-1 refractory cancers [110].
Mounting evidence demonstrates blocking one immune checkpoint can result in the upregulation of alternative modulators which potentially synergizes T cell exhaustion [111] and gives rise to compensatory mechanisms for immune evasion [111][112]. As a result of these studies, there are several antibodies against TIM-3 (e.g., TSR-022/Cobolimab, MBG453, LY3321367, BMS986258) being evaluated in clinical trials, mostly in combination with additional agents abrogating PD-1 and PD-L1 pathways [100]. Hence, future innovative BC treatment regimens may comprise TIM-3 co-blockage with additional checkpoint modulators, through the delivery of a blockade antibody cocktail or with miR-149-3p, in conjunction with non-immune-based protocols such as chemotherapy.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14020285
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Hammerl, D.; Smid, M.; Timmermans, A.M.; Sleijfer, S.; Martens, J.W.M.; Debets, R. Breast cancer genomics and immuno-oncological markers to guide immune therapies. Semin. Cancer Biol. 2018, 52, 178–188.
- Prat, A.; Cheang, M.C.; Martin, M.; Parker, J.S.; Carrasco, E.; Caballero, R.; Tyldesley, S.; Gelmon, K.; Bernard, P.S.; Nielsen, T.O.; et al. Prognostic significance of progesterone receptor-positive tumor cells within immunohistochemically defined luminal A breast cancer. J. Clin. Oncol. 2013, 31, 203–209.
- Ibrahim, E.; Al-Gahmi, A.M.; Zeenelin, A.A.; Zekri, J.M.; Elkhodary, T.R.; Gaballa, H.E.; Fawzy, E.E.; El Sayed, M.E.; Alzahrani, M.S. Basal vs. luminal A breast cancer subtypes: A matched case-control study using estrogen receptor, progesterone receptor, and HER-2 as surrogate markers. Med. Oncol. 2009, 26, 372–378.
- Mehta, R.J.; Jain, R.K.; Leung, S.; Choo, J.; Nielsen, T.; Huntsman, D.; Nakshatri, H.; Badve, S. FOXA1 is an independent prognostic marker for ER-positive breast cancer. Breast Cancer Res. Treat. 2012, 131, 881–890.
- Prat, A.; Pineda, E.; Adamo, B.; Galvan, P.; Fernandez, A.; Gaba, L.; Diez, M.; Viladot, M.; Arance, A.; Munoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. 2), S26–S35.
- Gadi, V.K.; Davidson, N.E. Practical Approach to Triple-Negative Breast Cancer. J. Oncol. Pract. 2017, 13, 293–300.
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes: A Systematic Review. JAMA Oncol. 2016, 2, 1354–1360.
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281.
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220.
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128.
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25.
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499.
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569.
- Fehres, C.M.; Unger, W.W.; Garcia-Vallejo, J.J.; van Kooyk, Y. Understanding the biology of antigen cross-presentation for the design of vaccines against cancer. Front. Immunol. 2014, 5, 149.
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198.
- Bates, J.P.; Derakhshandeh, R.; Jones, L.; Webb, T.J. Mechanisms of immune evasion in breast cancer. BMC Cancer 2018, 18, 556.
- Smyth, M.J.; Dunn, G.P.; Schreiber, R.D. Cancer immunosurveillance and immunoediting: The roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv. Immunol. 2006, 90, 1–50.
- Spranger, S.; Sivan, A.; Corrales, L.; Gajewski, T.F. Tumor and Host Factors Controlling Antitumor Immunity and Efficacy of Cancer Immunotherapy. Adv. Immunol. 2016, 130, 75–93.
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271.
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570.
- Forero, A.; Li, Y.; Chen, D.; Grizzle, W.E.; Updike, K.L.; Merz, N.D.; Downs-Kelly, E.; Burwell, T.C.; Vaklavas, C.; Buchsbaum, D.J.; et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 390–399.
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187.
- Wang, R.F.; Wang, H.Y. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res. 2017, 27, 11–37.
- de Marco, M.C.; Martin-Belmonte, F.; Kremer, L.; Albar, J.P.; Correas, I.; Vaerman, J.P.; Marazuela, M.; Byrne, J.A.; Alonso, M.A. MAL2, a novel raft protein of the MAL family, is an essential component of the machinery for transcytosis in hepatoma HepG2 cells. J. Cell Biol. 2002, 159, 37–44.
- Fang, Y.; Wang, L.; Wan, C.; Sun, Y.; Van der Jeught, K.; Zhou, Z.; Dong, T.; So, K.M.; Yu, T.; Li, Y.; et al. MAL2 drives immune evasion in breast cancer by suppressing tumor antigen presentation. J. Clin. Investig. 2021, 131, e140837.
- Vitale, M.; Rezzani, R.; Rodella, L.; Zauli, G.; Grigolato, P.; Cadei, M.; Hicklin, D.J.; Ferrone, S. HLA class I antigen and transporter associated with antigen processing (TAP1 and TAP2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res. 1998, 58, 737–742.
- Chen, H.L.; Gabrilovich, D.; Virmani, A.; Ratnani, I.; Girgis, K.R.; Nadaf-Rahrov, S.; Fernandez-Vina, M.; Carbone, D.P. Structural and functional analysis of beta2 microglobulin abnormalities in human lung and breast cancer. Int. J. Cancer 1996, 67, 756–763.
- Nomura, T.; Huang, W.C.; Zhau, H.E.; Josson, S.; Mimata, H.; Chung, L.W. beta2-Microglobulin-mediated signaling as a target for cancer therapy. Anticancer Agents Med. Chem. 2014, 14, 343–352.
- Kaneko, K.; Ishigami, S.; Kijima, Y.; Funasako, Y.; Hirata, M.; Okumura, H.; Shinchi, H.; Koriyama, C.; Ueno, S.; Yoshinaka, H.; et al. Clinical implication of HLA class I expression in breast cancer. BMC Cancer 2011, 11, 454.
- da Silva, G.B.; Silva, T.G.; Duarte, R.A.; Neto, N.L.; Carrara, H.H.; Donadi, E.A.; Goncalves, M.A.; Soares, E.G.; Soares, C.P. Expression of the Classical and Nonclassical HLA Molecules in Breast Cancer. Int. J. Breast Cancer 2013, 2013, 250435.
- Harada, A.; Ishigami, S.; Kijima, Y.; Nakajo, A.; Arigami, T.; Kurahara, H.; Kita, Y.; Yoshinaka, H.; Natsugoe, S. Clinical implication of human leukocyte antigen (HLA)-F expression in breast cancer. Pathol. Int. 2015, 65, 569–574.
- Konig, L.; Kasimir-Bauer, S.; Hoffmann, O.; Bittner, A.K.; Wagner, B.; Manvailer, L.F.; Schramm, S.; Bankfalvi, A.; Giebel, B.; Kimmig, R.; et al. The prognostic impact of soluble and vesicular HLA-G and its relationship to circulating tumor cells in neoadjuvant treated breast cancer patients. Hum. Immunol. 2016, 77, 791–799.
- De Kruijf, E.M.; Engels, C.C.; van de Water, W.; Bastiaannet, E.; Smit, V.T.; van de Velde, C.J.; Liefers, G.J.; Kuppen, P.J. Tumor immune subtypes distinguish tumor subclasses with clinical implications in breast cancer patients. Breast Cancer Res. Treat. 2013, 142, 355–364.
- Engels, C.C.; Fontein, D.B.; Kuppen, P.J.; de Kruijf, E.M.; Smit, V.T.; Nortier, J.W.; Liefers, G.J.; van de Velde, C.J.; Bastiaannet, E. Immunological subtypes in breast cancer are prognostic for invasive ductal but not for invasive lobular breast carcinoma. Br. J. Cancer 2014, 111, 532–538.
- Lin, A.; Yan, W.H. Human Leukocyte Antigen-G (HLA-G) Expression in Cancers: Roles in Immune Evasion, Metastasis and Target for Therapy. Mol. Med. 2015, 21, 782–791.
- Tao, S.; He, H.; Chen, Q.; Yue, W. GPER mediated estradiol reduces miR-148a to promote HLA-G expression in breast cancer. Biochem. Biophys. Res. Commun. 2014, 451, 74–78.
- Sheu, J.; Shih Ie, M. HLA-G and immune evasion in cancer cells. J. Formos. Med. Assoc. 2010, 109, 248–257.
- Garg, A.D.; More, S.; Rufo, N.; Mece, O.; Sassano, M.L.; Agostinis, P.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology 2017, 6, e1386829.
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966.
- Chatterjee, S.; Chatterjee, A.; Jana, S.; Dey, S.; Roy, H.; Das, M.K.; Alam, J.; Adhikary, A.; Chowdhury, A.; Biswas, A.; et al. Transforming growth factor beta orchestrates PD-L1 enrichment in tumor-derived exosomes and mediates CD8 T-cell dysfunction regulating early phosphorylation of TCR signalome in breast cancer. Carcinogenesis 2021, 42, 38–47.
- Yamashita, N.; Long, M.; Fushimi, A.; Yamamoto, M.; Hata, T.; Hagiwara, M.; Bhattacharya, A.; Hu, Q.; Wong, K.K.; Liu, S.; et al. MUC1-C integrates activation of the IFN-gamma pathway with suppression of the tumor immune microenvironment in triple-negative breast cancer. J. Immunother. Cancer 2021, 9, e002115.
- Steven, A.; Seliger, B. The Role of Immune Escape and Immune Cell Infiltration in Breast Cancer. Breast Care 2018, 13, 16–21.
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175.
- Bagati, A.; Kumar, S.; Jiang, P.; Pyrdol, J.; Zou, A.E.; Godicelj, A.; Mathewson, N.D.; Cartwright, A.N.R.; Cejas, P.; Brown, M.; et al. Integrin alphavbeta6-TGFbeta-SOX4 Pathway Drives Immune Evasion in Triple-Negative Breast Cancer. Cancer Cell 2021, 39, 54–67.e59.
- Mojic, M.; Takeda, K.; Hayakawa, Y. The Dark Side of IFN-gamma: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2017, 19, 89.
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847.
- Kufe, D.W. MUC1-C oncoprotein as a target in breast cancer: Activation of signaling pathways and therapeutic approaches. Oncogene 2013, 32, 1073–1081.
- Kufe, D.W. MUC1-C in chronic inflammation and carcinogenesis; emergence as a target for cancer treatment. Carcinogenesis 2020, 41, 1173–1183.
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811.
- Chikman, B.; Vasyanovich, S.; Lavy, R.; Habler, L.; Tolstov, G.; Kapiev, A.; Halevy, A.; Sandbank, J. COX2 expression in high-grade breast cancer: Evidence for prognostic significance in the subset of triple-negative breast cancer patients. Med. Oncol. 2014, 31, 989.
- Wu, A.A.; Drake, V.; Huang, H.S.; Chiu, S.; Zheng, L. Reprogramming the tumor microenvironment: Tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology 2015, 4, e1016700.
- Swoboda, A.; Nanda, R. Immune Checkpoint Blockade for Breast Cancer. Cancer Treat. Res. 2018, 173, 155–165.
- Michel, L.L.; von Au, A.; Mavratzas, A.; Smetanay, K.; Schutz, F.; Schneeweiss, A. Immune Checkpoint Blockade in Patients with Triple-Negative Breast Cancer. Target Oncol. 2020, 15, 415–428.
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126.
- Van Coillie, S.; Wiernicki, B.; Xu, J. Molecular and Cellular Functions of CTLA-4. Adv. Exp. Med. Biol. 2020, 1248, 7–32.
- Urbano, A.C.; Nascimento, C.; Soares, M.; Correia, J.; Ferreira, F. Clinical Relevance of the serum CTLA-4 in Cats with Mammary Carcinoma. Sci. Rep. 2020, 10, 3822.
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167.
- Muenst, S.; Soysal, S.D.; Gao, F.; Obermann, E.C.; Oertli, D.; Gillanders, W.E. The presence of programmed death 1 (PD-1)-positive tumor-infiltrating lymphocytes is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2013, 139, 667–676.
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370.
- Barrett, M.T.; Lenkiewicz, E.; Malasi, S.; Basu, A.; Yearley, J.H.; Annamalai, L.; McCullough, A.E.; Kosiorek, H.E.; Narang, P.; Wilson Sayres, M.A.; et al. The association of genomic lesions and PD-1/PD-L1 expression in resected triple-negative breast cancers. Breast Cancer Res. 2018, 20, 71.
- Muenst, S.; Schaerli, A.R.; Gao, F.; Daster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24.
- MuenstMaeda, T.; Hiraki, M.; Jin, C.; Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Hu, X.; Suzuki, Y.; Miyo, M.; et al. MUC1-C Induces PD-L1 and Immune Evasion in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 205–215.
- Yao, H.; Lan, J.; Li, C.; Shi, H.; Brosseau, J.P.; Wang, H.; Lu, H.; Fang, C.; Zhang, Y.; Liang, L.; et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat. Biomed. Eng. 2019, 3, 306–317.
- Nouri, K.; Azad, T.; Lightbody, E.; Khanal, P.; Nicol, C.J.; Yang, X. A kinome-wide screen using a NanoLuc LATS luminescent biosensor identifies ALK as a novel regulator of the Hippo pathway in tumorigenesis and immune evasion. FASEB J. 2019, 33, 12487–12499.
- Darvin, P.; Sasidharan Nair, V.; Elkord, E. PD-L1 Expression in Human Breast Cancer Stem Cells Is Epigenetically Regulated through Posttranslational Histone Modifications. J. Oncol. 2019, 2019, 3958908.
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060.
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep. 2016, 16, 2829–2837.
- Jing, X.; Shao, S.; Zhang, Y.; Luo, A.; Zhao, L.; Zhang, L.; Gu, S.; Zhao, X. BRD4 inhibition suppresses PD-L1 expression in triple-negative breast cancer. Exp. Cell Res. 2020, 392, 112034.
- Sasidharan Nair, V.; Toor, S.M.; Ali, B.R.; Elkord, E. Dual inhibition of STAT1 and STAT3 activation downregulates expression of PD-L1 in human breast cancer cells. Expert Opin. Ther. Targets 2018, 22, 547–557.
- Liu, J.; Yang, Y.; Wang, H.; Wang, B.; Zhao, K.; Jiang, W.; Bai, W.; Liu, J.; Yin, J. Syntenin1/MDA-9 (SDCBP) induces immune evasion in triple-negative breast cancer by upregulating PD-L1. Breast Cancer Res. Treat. 2018, 171, 345–357.
- Kumar, S.; Davra, V.; Obr, A.E.; Geng, K.; Wood, T.L.; De Lorenzo, M.S.; Birge, R.B. Crk adaptor protein promotes PD-L1 expression, EMT and immune evasion in a murine model of triple-negative breast cancer. Oncoimmunology 2017, 7, e1376155.
- Martinez, V.G.; O’Neill, S.; Salimu, J.; Breslin, S.; Clayton, A.; Crown, J.; O’Driscoll, L. Resistance to HER2-targeted anti-cancer drugs is associated with immune evasion in cancer cells and their derived extracellular vesicles. Oncoimmunology 2017, 6, e1362530.
- Timaner, M.; Kotsofruk, R.; Raviv, Z.; Magidey, K.; Shechter, D.; Kan, T.; Nevelsky, A.; Daniel, S.; de Vries, E.G.E.; Zhang, T.; et al. Microparticles from tumors exposed to radiation promote immune evasion in part by PD-L1. Oncogene 2020, 39, 187–203.
- Zam, W.; Ali, L. Immune checkpoint inhibitors in the treatment of cancer. Curr. Clin. Pharmacol. 2021.
- Alshehri, B. Immunotherapy: New insights in breast cancer treatment. Hum. Antib. 2021, 29, 193–202.
- Kwapisz, D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 2021, 70, 607–617.
- Terranova-Barberio, M.; Thomas, S.; Ali, N.; Pawlowska, N.; Park, J.; Krings, G.; Rosenblum, M.D.; Budillon, A.; Munster, P.N. HDAC inhibition potentiates immunotherapy in triple negative breast cancer. Oncotarget 2017, 8, 114156–114172.
- Olino, K.; Park, T.; Ahuja, N. Exposing Hidden Targets: Combining epigenetic and immunotherapy to overcome cancer resistance. Semin. Cancer Biol. 2020, 65, 114–122.
- Force, J.; Leal, J.H.S.; McArthur, H.L. Checkpoint Blockade Strategies in the Treatment of Breast Cancer: Where We Are and Where We Are Heading. Curr. Treat. Options Oncol. 2019, 20, 35.
- Kang, J.; Demaria, S.; Formenti, S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J. Immunother. Cancer 2016, 4, 51.
- Pieper, A.A.; Rakhmilevich, A.L.; Spiegelman, D.V.; Patel, R.B.; Birstler, J.; Jin, W.J.; Carlson, P.M.; Charych, D.H.; Hank, J.A.; Erbe, A.K.; et al. Combination of radiation therapy, bempegaldesleukin, and checkpoint blockade eradicates advanced solid tumors and metastases in mice. J. Immunother. Cancer 2021, 9, e002715.
- Theelen, W.; Chen, D.; Verma, V.; Hobbs, B.P.; Peulen, H.M.U.; Aerts, J.; Bahce, I.; Niemeijer, A.L.N.; Chang, J.Y.; de Groot, P.M.; et al. Pembrolizumab with or without radiotherapy for metastatic non-small-cell lung cancer: A pooled analysis of two randomised trials. Lancet Respir. Med. 2021, 9, 467–475.
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405.
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337.
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721.
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499.
- Schnorfeil, F.M.; Lichtenegger, F.S.; Emmerig, K.; Schlueter, M.; Neitz, J.S.; Draenert, R.; Hiddemann, W.; Subklewe, M. T cells are functionally not impaired in AML: Increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J. Hematol. Oncol. 2015, 8, 93.
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880.
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927.
- Lichtenegger, F.S.; Rothe, M.; Schnorfeil, F.M.; Deiser, K.; Krupka, C.; Augsberger, C.; Schluter, M.; Neitz, J.; Subklewe, M. Targeting LAG-3 and PD-1 to Enhance T Cell Activation by Antigen-Presenting Cells. Front. Immunol. 2018, 9, 385.
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423.
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312.
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541.
- Yasinska, I.M.; Sakhnevych, S.S.; Pavlova, L.; Teo Hansen Selno, A.; Teuscher Abeleira, A.M.; Benlaouer, O.; Goncalves Silva, I.; Mosimann, M.; Varani, L.; Bardelli, M.; et al. The Tim-3-Galectin-9 Pathway and Its Regulatory Mechanisms in Human Breast Cancer. Front. Immunol. 2019, 10, 1594.
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252.
- Sabatos-Peyton, C.A.; Nevin, J.; Brock, A.; Venable, J.D.; Tan, D.J.; Kassam, N.; Xu, F.; Taraszka, J.; Wesemann, L.; Pertel, T.; et al. Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-Tim-3 antibodies that have functional efficacy. Oncoimmunology 2018, 7, e1385690.
- Fang, J.; Chen, F.; Liu, D.; Gu, F.; Chen, Z.; Wang, Y. Prognostic value of immune checkpoint molecules in breast cancer. Biosci. Rep. 2020, 40, BSR20201054.
- de Mingo Pulido, A.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74 e66.
- de Mingo Pulido, A.; Hanggi, K.; Celias, D.P.; Gardner, A.; Li, J.; Batista-Bittencourt, B.; Mohamed, E.; Trillo-Tinoco, J.; Osunmakinde, O.; Pena, R.; et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 2021, 54, 1154–1167 e1157.
- Gardner, A.; de Mingo Pulido, A.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924.
- Cong, Y.; Cui, Y.; Zhu, S.; Cao, J.; Zou, H.; Martin, T.A.; Qiao, G.; Jiang, W.; Yu, Z. Tim-3 promotes cell aggressiveness and paclitaxel resistance through NF-kappaB/STAT3 signalling pathway in breast cancer cells. Chin. J. Cancer Res. 2020, 32, 564–579.
- Liu, Q.; Yu, S.; Li, A.; Xu, H.; Han, X.; Wu, K. Targeting interlukin-6 to relieve immunosuppression in tumor microenvironment. Tumour. Biol. 2017, 39, 1010428317712445.
- Wang, Y.; Shen, Y.; Wang, S.; Shen, Q.; Zhou, X. The role of STAT3 in leading the crosstalk between human cancers and the immune system. Cancer Lett. 2018, 415, 117–128.
- Cheng, S.; Han, F.; Xu, Y.; Qu, T.; Ju, Y. Expression of Tim-3 in breast cancer tissue promotes tumor progression. Int. J. Clin. Exp. Pathol. 2018, 11, 1157–1166.
- Zhang, M.; Gao, D.; Shi, Y.; Wang, Y.; Joshi, R.; Yu, Q.; Liu, D.; Alotaibi, F.; Zhang, Y.; Wang, H.; et al. miR-149-3p reverses CD8(+) T-cell exhaustion by reducing inhibitory receptors and promoting cytokine secretion in breast cancer cells. Open Biol. 2019, 9, 190061.
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911.
- Ngiow, S.F.; von Scheidt, B.; Akiba, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011, 71, 3540–3551.
- Klein, C.; Schaefer, W.; Regula, J.T.; Dumontet, C.; Brinkmann, U.; Bacac, M.; Umana, P. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods 2019, 154, 21–31.
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501.
- Saleh, R.; Toor, S.M.; Khalaf, S.; Elkord, E. Breast Cancer Cells and PD-1/PD-L1 Blockade Upregulate the Expression of PD-1, CTLA-4, TIM-3 and LAG-3 Immune Checkpoints in CD4(+) T Cells. Vaccines 2019, 7, 149.
- Saleh, R.; Elkord, E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. 2019, 457, 168–179.
This entry is offline, you can click here to edit this entry!