One-carbon metabolism (OCM) is a network of biochemical reactions delivering one-carbon units to various biosynthetic pathways. The folate cycle and methionine cycle are the two key modules of this network that regulate purine and thymidine synthesis, amino acid homeostasis, and epigenetic mechanisms. Intersection with the transsulfuration pathway supports glutathione production and regulation of the cellular redox state. Dietary intake of micronutrients, such as folates and amino acids, directly contributes to OCM, thereby adapting the cellular metabolic state to environmental inputs. The contribution of OCM to cellular proliferation during development and in adult proliferative tissues is well established. Nevertheless, accumulating evidence reveals the pivotal role of OCM in cellular homeostasis of non-proliferative tissues and in coordination of signaling cascades that regulate energy homeostasis and longevity.
- aging
- Alzheimer’s disease
- diet
- folate
- metabolism
- methionine
- mitochondria
- neurodegeneration
- one-carbon vitamins
- Parkinson disease
1. Introduction
2. One-Carbon Metabolic Pathways
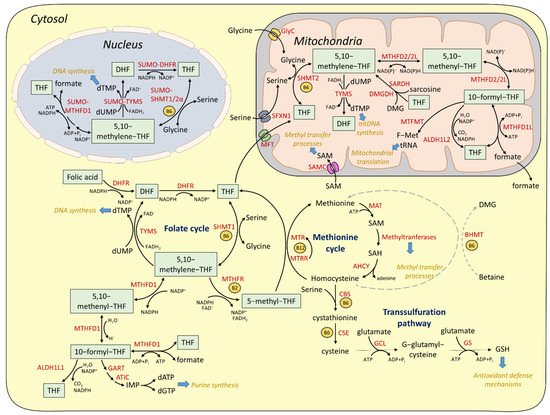
2.1. Cytosolic and Nuclear Pathways of One-Carbon Metabolism
2.2. Mitochondrial One-Carbon Metabolism
3. One-Carbon Metabolism in Aging
| Genetic Manipulation | Environmental Manipulation | Effect on Lifespan | References |
|---|---|---|---|
| Yeast | |||
| Methionine restriction | CLS extension | [54,55] | |
| met2 deletion | CLS extension | [56,57] | |
| met15 deletion | CLS extension | [56,57] | |
| met3 deletion | RLS extension | [58] | |
| sam1 deletion | RLS extension | [58] | |
| cys4 deletion | CLS extension | [59] | |
| H2S | CLS extension | [60] | |
| gsh1 deletion | 10% glucose | CLS shortening | [61] |
| Nematodes | |||
| sulfamethoxazole | Extension | [62] | |
| dhfr-1 RNAi | extension | [63] | |
| tyms-1 RNAi | extension | [63,64] | |
| daf-2(e1370) | MTHF5 supplementation | Reverses longevity of daf-2 mutants |
[63] |
| mel-32 RNAi | extension | [65] | |
| sams-1 RNAi | extension | [66,67] | |
| sams-5 RNAi | extension | [67] | |
| Glycine supplementation | extension | [65,68] | |
| Serine supplementation | extension | [65,68] | |
| Metformin | extension | [69] | |
| sams-1 mutant | Metformin | Reverse of metformin’s benefits | [69] |
| metr-1 mutant | Metformin | Reverse of metformin’s benefits | [69] |
| H2S | extension | [70,71] | |
| cbs-1 overexpression | extension | [60] | |
| NAC from day 3 | extension | [72] | |
| NAC from L4 | shortening | [73] | |
| taurine | extension | [68] | |
| Acivicin (GSH restriction) | extension | [73] | |
| Flies | |||
| GNMT overexpression | extension | [74] | |
| Methionine restriction | extension | [75,76] | |
| Sams depletion | extension | [74] | |
| Cbs overexpression | extension | [77] | |
| Cbs depletion | Caloric restriction | Reversed CR-driven longevity | [78] |
| Gclc overexpression | extension | [79] | |
| Gclm overexpression | extension | [79] | |
| dAhcyL1/dAhcyL2 suppression | Extension | [80] | |
| Mammals | |||
| Transgenic growth hormone mice | Supplementation of several forms of folate | Extension | [81] |
| wt | Supplementation of several forms of folate | Extension | [81] |
| Female SHR mice | metformin | Extension | [82] |
| Rats, mice | Dietary Methionine restriction | Extension | [83,84,85] |
| Tissue-specific taurine transporter depleted mice | Shortening | [86] | |
| Humans | |||
| C667T MTHFR | Associates with decrease in all-cause mortality | [87,88] | |
3.1. Folate Cycle in Aging
3.2. Methionine Cycle in Aging
3.3. Homocysteine in Aging
3.4. Transsulfuration Pathway in Aging
3.5. Glutathione in Aging
4. One-Carbon Metabolism and Neurodegeneration
4.1. One-Carbon Metabolism in the Pathogenesis of Alzheimer’s Disease
4.2. One-Carbon Metabolism in the Pathogenesis of Parkinson’s Disease
This entry is adapted from the peer-reviewed paper 10.3390/cells11020214