The main protease (M-pro) or 3C-like protease of coronaviruses plays an essential role in virus replication. This protease contributes to the cleavage of the ppa1a and pp1ab polyproteins to produce several non-structural proteins, including M-pro itself. Since the beginning of the COVID-19 pandemic, the SARS-CoV-2 M-pro enzyme has been extensively studied, and its inhibitors are promising effective drugs for fighting against SARS-CoV-2. The first attempts to discover SARS-CoV-2 M-pro inhibitors used previously developed protease inhibitors or tried to repurpose drugs from other diseases. Covalent inhibitors form a covalent bond, usually with catalytic Cys145. Non-covalent inhibitors bind by non-covalent interactions at the active site of the enzyme, inhibiting its function.
- COVID-19
- M-pro inhibitors
- 3CL-pro inhibitors
- computational chemistry
- protease inhibitors
- virtual screening
1. Introduction
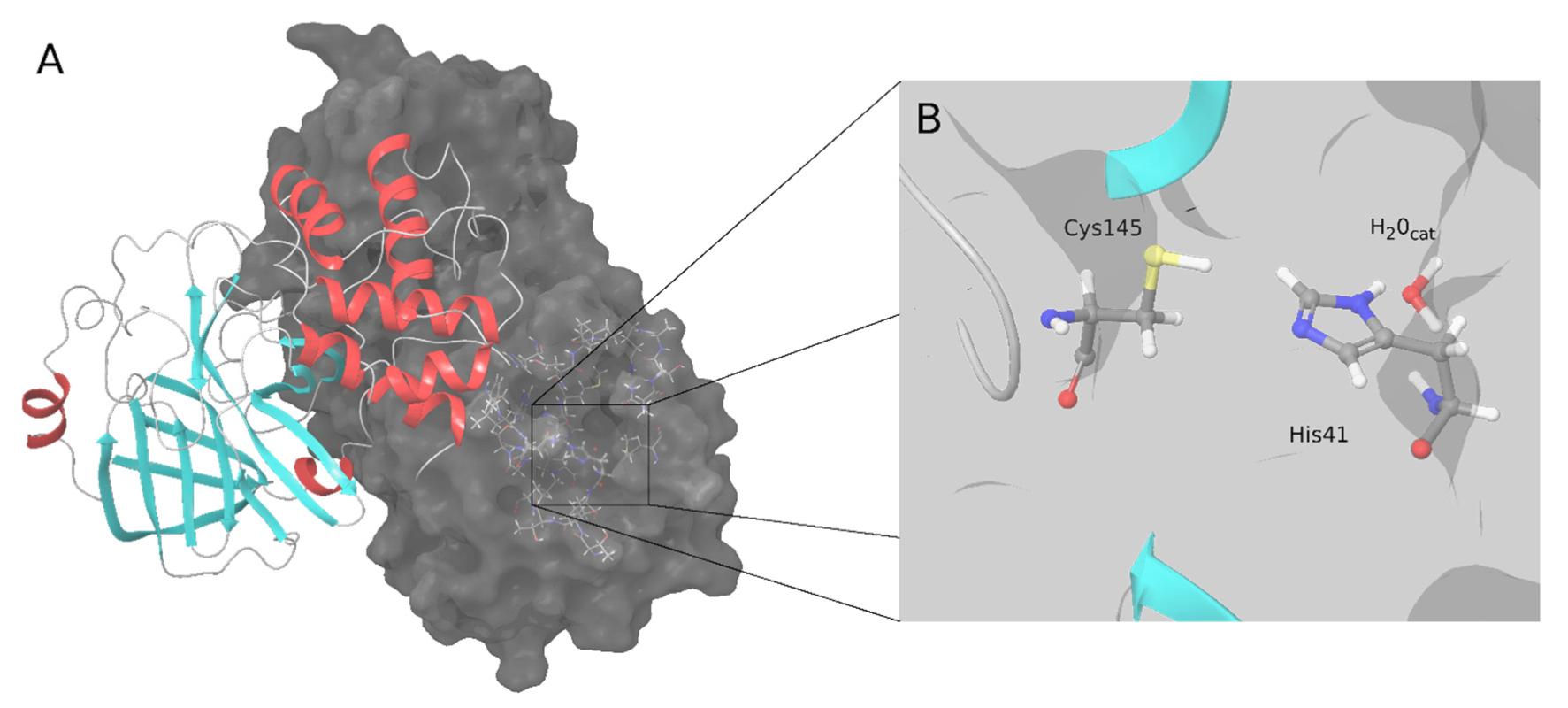
2. SARS-CoV-2 M-Pro Inhibitors
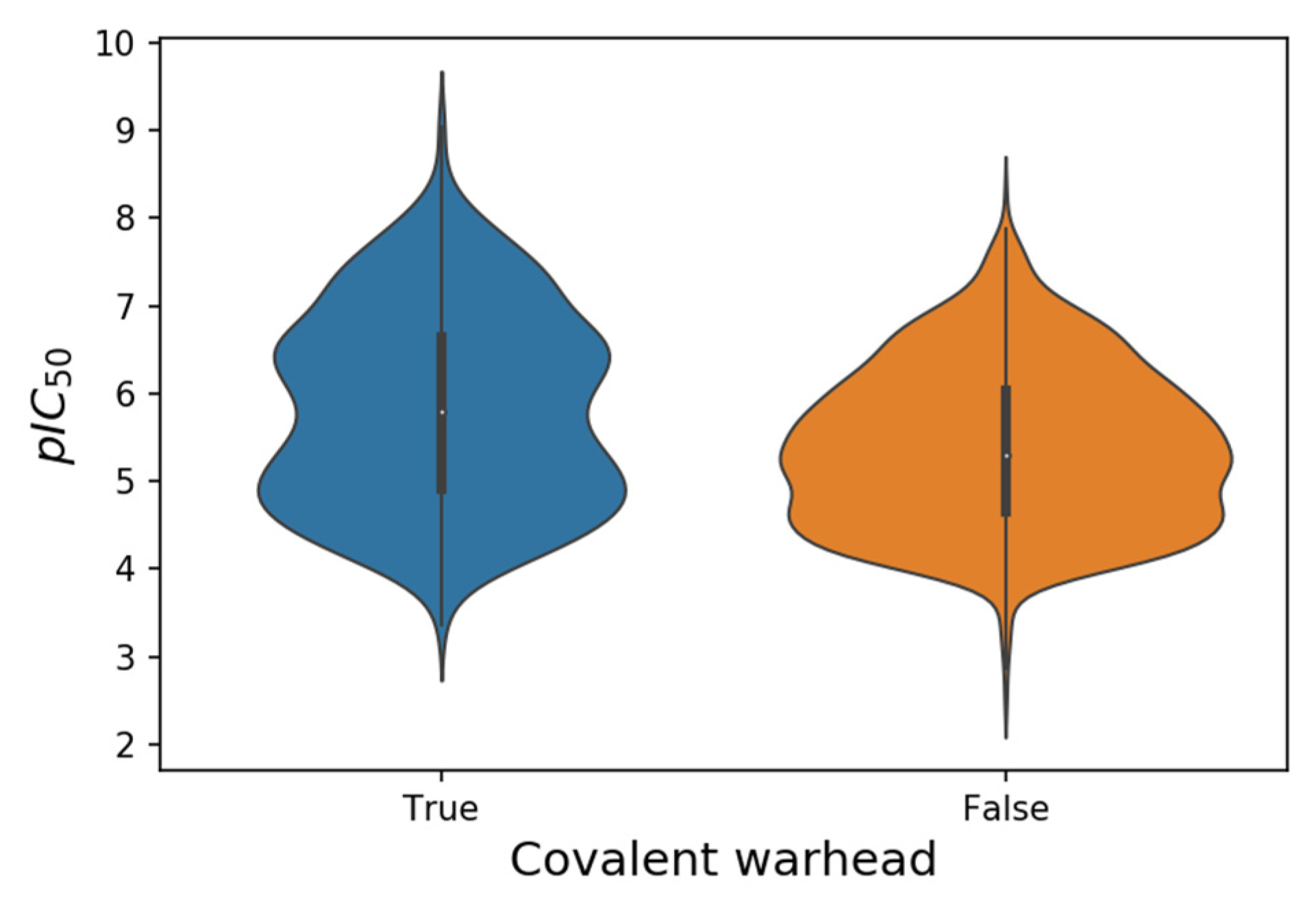
| SARS-CoV-2 M-Pro Inhibitor Set | Number of Compounds (Covalent/Non-Covalent) 1 | pIC50 Range | pIC50 Range Covalent | pIC50 Range Non-Covalent |
|---|---|---|---|---|
| From the bibliography | 758 (346/412) | 2.5–9.0 | 3.4–9.0 | 2.5–8.3 |
| From COVID Moonshot | 999 (205/794) | 4.0–7.8 | 4.0–7.8 | 4.0–7.4 |
| From ChEMBL | 8 (1/7) | 5.4–6.1 | 5.4 | 5.5–6.1 |
| All | 1765 (552/1213) | 2.5–9.0 | 3.4–9.0 | 2.5–8.3 |
| Warhead | SMARTS | Examples |
|---|---|---|
| Acrylamide | [C;H2:1]=[C;H1]C(N)=O | CVD-0004255 |
| Chloroacetamide | Cl[C;H2:1]C(N)=O | BFC204 |
| Vinylsulfonamide | NS(=O)([C;H1]=[C;H2:1])=O | |
| Nitrile | N#[C:1]-[*] | Isavuconazole |
| Michael acceptors | C=!@CC=[O,S] | Cinanserin, MPI2, MPI9, N3 |
| Alpha-ketoamide | C(=O)(C=O)N | Boceprevir, narlaprevir, telaprevir, UAWJ248 |
| Aldehyde | [CX3H1](=O) | GC373, MI-05, MI-06, MI-09, MI-11, MI-13, MI-14, MI-21, MI-23, MI-28 |
| Bisulfite adduct of aldehyde | C(O)S(=[OX1])([O])(=[OX1]) | GC376 |
| Urea carbonyl | [NX3][CX3](=[OX1])([NX3,nX3]) | Carmofur |
| Bis(dialkylaminethiocarbonyl)disulfide | [CX3](=[SX1])SS[CX3](=[SX1]) | Disulfiram |
| Carbamoylsulfanyl | [NX3,nX3][C,c](=[OX1])([SX2,sx2]) | Tideglusib |
| Disulfide | [SX2][SX2] | PX-12 |
| Hydroxymethyl ketone | [CX3H0](=[OX1])[CH2][OH] | PF-00835231 |
| Alkoxymethyl ketone | [CX3H0](=[OX1])[CH2][OX2H0] | 2683066-41-1, 2683066-42-2, 2683066-47-7 |
| Acyloxymethyl ketone | [CX3H0](=[OX1])[CH2][OX2H0][CX3H0](=O) | 2683066-41-1, 2683066-42-2, 2683066-47-7 |
| Fluoro, Chloro-methyl ketone | [CX3H0](=[OX1])[CH2][Cl,F] | Z-AVLD-FMK |
| Ebselen related | [Se]n(c=O) | Ebselen |
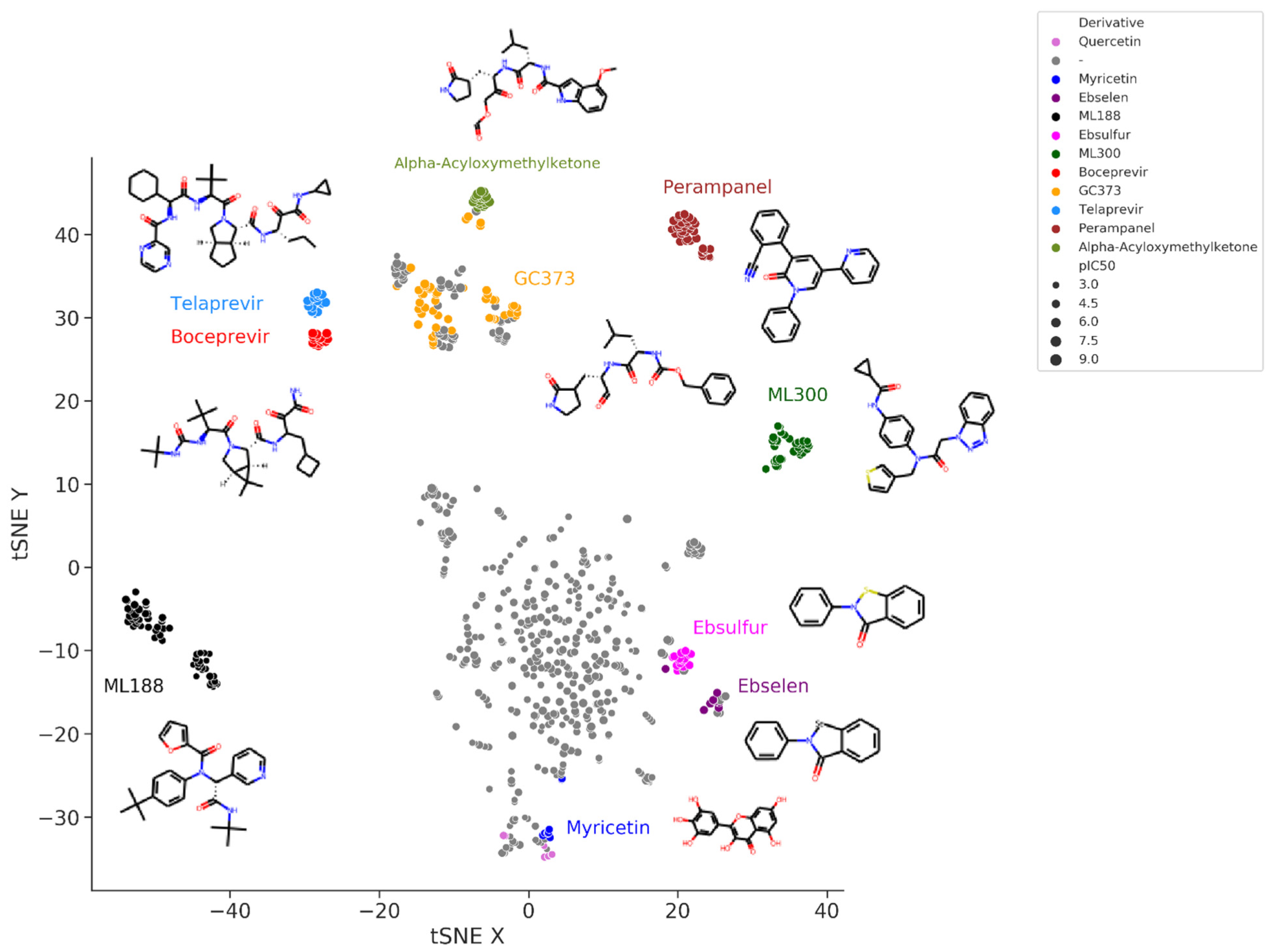
3. Conclusions
Although we have not yet hit the bullseye and no drug has yet been approved to inhibit SARS-CoV-2 M-pro, we may be close. Improving derivatives of a leading compound has proven to be a very successful strategy for finding potent SARS-CoV-2 M-pro inhibitors. Some derivative compounds designed in less than two years since the start of the COVID-19 pandemic represent an important step toward the development of new anti–SARS-CoV-2 drugs. Currently, there are several compounds with low nanomolar IC50 values against SARS-CoV-2 M-pro and high anti-SARS-CoV-2 efficacy in cell models, with values comparable to those of the FDA-approved RNA polymerase inhibitor remdesivir.This entry is adapted from the peer-reviewed paper 10.3390/ijms23010259
References
- Ziebuhr, J.; Herold, J.; Siddell, S.G. Characterization of a human coronavirus (strain 229E) 3C-like proteinase activity. J. Virol. 1995, 69, 4331–4338.
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of Novel Inhibitors of the Main Protease (M-pro) of SARS-CoV-2 through Consensus Docking and Drug Reposition. Int. J. Mol. Sci. 2020, 21, 3793.
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767.
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293.
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 3202.
- El-Baba, T.J.; Lutomski, C.A.; Kantsadi, A.L.; Malla, T.R.; John, T.; Mikhailov, V.; Bolla, J.R.; Schofield, C.J.; Zitzmann, N.; Vakonakis, I.; et al. Allosteric Inhibition of the SARS-CoV-2 Main Protease: Insights from Mass Spectrometry Based Assays. Angew. Chem. Int. Ed. Engl. 2020, 59, 23544–23548.
- Günther, S.; Reinke, P.Y.A.; Fernández-García, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 2021, 372, 642–646.
- Du, R.; Cooper, L.; Chen, Z.; Lee, H.; Rong, L.; Cui, Q. Discovery of chebulagic acid and punicalagin as novel allosteric inhibitors of SARS-CoV-2 3CLpro. Antivir. Res. 2021, 190, 105075.
- Eberle, R.J.; Olivier, D.S.; Amaral, M.S.; Gering, I.; Willbold, D.; Arni, R.K.; Coronado, M.A. The Repurposed Drugs Suramin and Quinacrine Cooperatively Inhibit SARS-CoV-2 3CLpro In Vitro. Viruses 2021, 13, 873.
- Nguyen, D.D.; Gao, K.; Chen, J.; Wang, R.; Wei, G.-W. Unveiling the molecular mechanism of SARS-CoV-2 main protease inhibition from 137 crystal structures using algebraic topology and deep learning. Chem. Sci. 2020, 11, 12036–12046.
- Liu, Z.; Fang, H.; Reagan, K.; Xu, X.; Mendrick, D.L.; Slikker, W.; Tong, W. In silico drug repositioning: What we need to know. Drug Discov. Today 2013, 18, 110–115.
- Chakraborti, S.; Bheemireddy, S.; Srinivasan, N. Repurposing drugs against the main protease of SARS-CoV-2: Mechanism-based insights supported by available laboratory and clinical data. Mol. Omics 2020, 16, 474–491.
- Wang, X.; Guan, Y. COVID-19 drug repurposing: A review of computational screening methods, clinical trials, and protein interaction assays. Med. Res. Rev. 2021, 41, 5–28.
- Gimeno, A.; Ojeda-Montes, M.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375.
- Warren, G.L.; Andrews, C.W.; Capelli, A.-M.; Clarke, B.; LaLonde, J.; Lambert, M.H.; Lindvall, M.; Nevins, N.; Semus, S.F.; Senger, S.; et al. A Critical Assessment of Docking Programs and Scoring Functions. J. Med. Chem. 2006, 49, 5912–5931.
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899.
- Bzówka, M.; Mitusińska, K.; Raczyńska, A.; Samol, A.; Tuszyński, J.A.; Góra, A. Structural and Evolutionary Analysis Indicate That the SARS-CoV-2 Mpro Is a Challenging Target for Small-Molecule Inhibitor Design. Int. J. Mol. Sci. 2020, 21, 3099.
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461.
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massagué, A.; Garcia-Vallvé, S.; Pujadas, G. Haste makes waste: A critical review of docking-based virtual screening in drug repurposing for SARS-CoV-2 main protease (M-pro) inhibition. Med. Res. Rev. 2021.
- Zev, S.; Raz, K.; Schwartz, R.; Tarabeh, R.; Gupta, P.K.; Major, D.T. Benchmarking the Ability of Common Docking Programs to Correctly Reproduce and Score Binding Modes in SARS-CoV-2 Protease Mpro. J. Chem. Inf. Model. 2021, 61, 2957–2966.
- Dotolo, S.; Marabotti, A.; Facchiano, A.; Tagliaferri, R. A review on drug repurposing applicable to COVID-19. Brief. Bioinform. 2021, 22, 726–741.
- Bellera, C.L.; Llanos, M.; Gantner, M.E.; Rodriguez, S.; Gavernet, L.; Comini, M.; Talevi, A. Can drug repurposing strategies be the solution to the COVID-19 crisis? Expert Opin. Drug Discov. 2021, 16, 605–612.
- Llanos, M.A.; Gantner, M.E.; Rodriguez, S.; Alberca, L.N.; Bellera, C.L.; Talevi, A.; Gavernet, L. Strengths and Weaknesses of Docking Simulations in the SARS-CoV-2 Era: The Main Protease (Mpro) Case Study. J. Chem. Inf. Model. 2021, 61, 3758–3770.
- Amin, S.A.; Banerjee, S.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery: What we have learned from the past SARS-CoV inhibitors? Eur. J. Med. Chem. 2021, 215, 113294.
- Yang, H.; Yang, J. A review of the latest research on Mpro targeting SARS-COV inhibitors. RSC Med. Chem. 2021, 12, 1026–1036.
- Cui, W.; Yang, K.; Yang, H. Recent Progress in the Drug Development Targeting SARS-CoV-2 Main Protease as Treatment for COVID-19. Front. Mol. Biosci. 2020, 7, 616341.
- Pillaiyar, T.; Wendt, L.L.; Manickam, M.; Easwaran, M. The recent outbreaks of human coronaviruses: A medicinal chemistry perspective. Med. Res. Rev. 2021, 41, 72–135.
- Banerjee, R.; Perera, L.; Tillekeratne, L.M.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discov. Today 2021, 26, 804–816.
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269.
- Gao, K.; Wang, R.; Chen, J.; Tepe, J.J.; Huang, F.; Wei, G.-W. Perspectives on SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2021, 64, 16922–16955.
- Xu, J.; Xue, Y.; Zhou, R.; Shi, P.; Li, H.; Zhou, J. Drug repurposing approach to combating coronavirus: Potential drugs and drug targets. Med. Res. Rev. 2021, 41, 1375–1426.
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 2020.
- Chen, C.; Yu, X.; Kuo, C.; Min, J.; Chen, S.; Ma, L.; Liu, K.; Guo, R. Overview of antiviral drug candidates targeting coronaviral 3C-like main proteases. FEBS J. 2021, 288, 5089–5121.
- Xiong, M.; Su, H.; Zhao, W.; Xie, H.; Shao, Q.; Xu, Y. What coronavirus 3C-like protease tells us: From structure, substrate selectivity, to inhibitor design. Med. Res. Rev. 2021, 41, 1965–1998.
- Mengist, H.M.; Dilnessa, T.; Jin, T. Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 2021, 9, 622898.
- Citarella, A.; Scala, A.; Piperno, A.; Micale, N. SARS-CoV-2 Mpro: A Potential Target for Peptidomimetics and Small-Molecule Inhibitors. Biomolecules 2021, 11, 607.
- Chia, C.S.B.; Xu, W.; Shuyi Ng, P. A Patent Review on SARS Coronavirus Main Protease (3CL pro) Inhibitors. ChemMedChem 2021.
- Vandyck, K.; Deval, J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr. Opin. Virol. 2021, 49, 36–40.
- COVID Moonshot. Available online: https://covid.postera.ai/covid (accessed on 1 October 2021).
- The COVID Moonshot Consortium; Chodera, J.; Lee, A.; London, N.; von Delft, F. Open Science Discovery of Oral Non-Covalent SARS-CoV-2 Main Protease Inhibitors. ChemRxiv 2021.
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940.
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317.
- Ghahremanpour, M.M.; Tirado-Rives, J.; Deshmukh, M.; Ippolito, J.A.; Zhang, C.-H.; Cabeza de Vaca, I.; Liosi, M.-E.; Anderson, K.S.; Jorgensen, W.L. Identification of 14 Known Drugs as Inhibitors of the Main Protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020, 11, 2526–2533.
- Zhang, C.-H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475.
- Deshmukh, M.G.; Ippolito, J.A.; Zhang, C.-H.; Stone, E.A.; Reilly, R.A.; Miller, S.J.; Jorgensen, W.L.; Anderson, K.S. Structure-guided design of a perampanel-derived pharmacophore targeting the SARS-CoV-2 main protease. Structure 2021, 29, 823–833.e5.
- Zhang, C.-H.; Spasov, K.A.; Reilly, R.A.; Hollander, K.; Stone, E.A.; Ippolito, J.A.; Liosi, M.-E.; Deshmukh, M.G.; Tirado-Rives, J.; Zhang, S.; et al. Optimization of Triarylpyridinone Inhibitors of the Main Protease of SARS-CoV-2 to Low-Nanomolar Antiviral Potency. ACS Med. Chem. Lett. 2021, 12, 1325–1332.
- Jacobs, J.; Grum-Tokars, V.; Zhou, Y.; Turlington, M.; Saldanha, S.A.; Chase, P.; Eggler, A.; Dawson, E.S.; Baez-Santos, Y.M.; Tomar, S.; et al. Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL pr. J. Med. Chem. 2013, 56, 534–546.
- Turlington, M.; Chun, A.; Tomar, S.; Eggler, A.; Grum-Tokars, V.; Jacobs, J.; Daniels, J.S.; Dawson, E.; Saldanha, A.; Chase, P.; et al. Discovery of N-(benzotriazol-1-yl)-N-(benzyl)acetamido)phenyl) carboxamides as severe acute respiratory syndrome coronavirus (SARS-CoV) 3CLpro inhibitors: Identification of ML300 and noncovalent nanomolar inhibitors with an induced-fit binding. Bioorg. Med. Chem. Lett. 2013, 23, 6172–6177.
- Han, S.H.; Goins, C.M.; Arya, T.; Shin, W.-J.; Maw, J.; Hooper, A.; Sonawane, D.P.; Porter, M.R.; Bannister, B.E.; Crouch, R.D.; et al. Structure-Based Optimization of ML300-Derived, Noncovalent Inhibitors Targeting the Severe Acute Respiratory Syndrome Coronavirus 3CL Protease (SARS-CoV-2 3CL pro). J. Med. Chem. 2021.
- Kitamura, N.; Sacco, M.D.; Ma, C.; Hu, Y.; Townsend, J.A.; Meng, X.; Zhang, F.; Zhang, X.; Ba, M.; Szeto, T.; et al. Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2021.
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692.
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417.
- Kneller, D.W.; Galanie, S.; Phillips, G.; O’Neill, H.M.; Coates, L.; Kovalevsky, A. Malleability of the SARS-CoV-2 3CL Mpro Active-Site Cavity Facilitates Binding of Clinical Antivirals. Structure 2020, 28, 1313–1320.e3.
- Pathak, N.; Chen, Y.-T.; Hsu, Y.-C.; Hsu, N.-Y.; Kuo, C.-J.; Tsai, H.P.; Kang, J.-J.; Huang, C.-H.; Chang, S.-Y.; Chang, Y.-H.; et al. Uncovering Flexible Active Site Conformations of SARS-CoV-2 3CL Proteases through Protease Pharmacophore Clusters and COVID-19 Drug Repurposing. ACS Nano 2021, 15, 857–872.
- Jan, J.-T.; Cheng, T.-J.R.; Juang, Y.-P.; Ma, H.-H.; Wu, Y.-T.; Yang, W.-B.; Cheng, C.-W.; Chen, X.; Chou, T.-H.; Shie, J.-J.; et al. Identification of existing pharmaceuticals and herbal medicines as inhibitors of SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2021579118.
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93.
- Baker, J.D.; Uhrich, R.L.; Kraemer, G.C.; Love, J.E.; Kraemer, B.C. A drug repurposing screen identifies hepatitis C antivirals as inhibitors of the SARS-CoV2 main protease. PLoS ONE 2021, 16, e0245962.
- Manandhar, A.; Blass, B.E.; Colussi, D.J.; Almi, I.; Abou-Gharbia, M.; Klein, M.L.; Elokely, K.M. Targeting SARS-CoV-2 M3CLpro by HCV NS3/4a Inhibitors: In Silico Modeling and In Vitro Screening. J. Chem. Inf. Model. 2021, 61, 1020–1032.
- Qiao, J.; Li, Y.-S.; Zeng, R.; Liu, F.-L.; Luo, R.-H.; Huang, C.; Wang, Y.-F.; Zhang, J.; Quan, B.; Shen, C.; et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 2021, 371, 1374–1378.
- Kim, Y.; Shivanna, V.; Narayanan, S.; Prior, A.M.; Weerasekara, S.; Hua, D.H.; Kankanamalage, A.C.G.; Groutas, W.C.; Chang, K.-O. Broad-spectrum inhibitors against 3C-like proteases of feline coronaviruses and feline caliciviruses. J. Virol. 2015, 89, 4942–4950.
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282.
- Hung, H.; Ke, Y.; Huang, S.Y.; Huang, P.-N.; Kung, Y.; Chang, T.-Y.; Yen, K.; Peng, T.-T.; Chang, S.-E.; Huang, C.-T.; et al. Discovery of M Protease Inhibitors Encoded by SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, e00872-20.
- Rathnayake, A.D.; Zheng, J.; Kim, Y.; Perera, K.D.; Mackin, S.; Meyerholz, D.K.; Kashipathy, M.M.; Battaile, K.P.; Lovell, S.; Perlman, S.; et al. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV–infected mice. Sci. Transl. Med. 2020, 12, eabc5332.
- Gurard-Levin, Z.A.; Liu, C.; Jekle, A.; Jaisinghani, R.; Ren, S.; Vandyck, K.; Jochmans, D.; Leyssen, P.; Neyts, J.; Blatt, L.M.; et al. Evaluation of SARS-CoV-2 3C-like protease inhibitors using self-assembled monolayer desorption ionization mass spectrometry. Antivir. Res. 2020, 182, 104924.
- Wang, Y.-C.; Yang, W.-H.; Yang, C.-S.; Hou, M.-H.; Tsai, C.-L.; Chou, Y.-Z.; Hung, M.-C.; Chen, Y. Structural basis of SARS-CoV-2 main protease inhibition by a broad-spectrum anti-coronaviral drug. Am. J. Cancer Res. 2020, 10, 2535–2545.
- Zhu, W.; Xu, M.; Chen, C.Z.; Guo, H.; Shen, M.; Hu, X.; Shinn, P.; Klumpp-Thomas, C.; Michael, S.G.; Zheng, W. Identification of SARS-CoV-2 3CL Protease Inhibitors by a Quantitative High-Throughput Screening. ACS Pharmacol. Transl. Sci. 2020, 3, 1008–1016.
- Iketani, S.; Forouhar, F.; Liu, H.; Hong, S.J.; Lin, F.-Y.; Nair, M.S.; Zask, A.; Huang, Y.; Xing, L.; Stockwell, B.R.; et al. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun. 2021, 12, 2016.
- Yang, K.S.; Ma, X.R.; Ma, Y.; Alugubelli, Y.R.; Scott, D.A.; Vatansever, E.C.; Drelich, A.K.; Sankaran, B.; Geng, Z.Z.; Blankenship, L.R.; et al. A Quick Route to Multiple Highly Potent SARS-CoV-2 Main Protease Inhibitors. ChemMedChem 2021, 16, 942–948.
- Vuong, W.; Fischer, C.; Khan, M.B.; van Belkum, M.J.; Lamer, T.; Willoughby, K.D.; Lu, J.; Arutyunova, E.; Joyce, M.A.; Saffran, H.A.; et al. Improved SARS-CoV-2 Mpro inhibitors based on feline antiviral drug GC376: Structural enhancements, increased solubility, and micellar studies. Eur. J. Med. Chem. 2021, 222, 113584.
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020, 6, eabe0751.
- Bai, B.; Belovodskiy, A.; Hena, M.; Kandadai, A.S.; Joyce, M.A.; Saffran, H.A.; Shields, J.A.; Khan, M.B.; Arutyunova, E.; Lu, J.; et al. Peptidomimetic α-Acyloxymethylketone Warheads with Six-Membered Lactam P1 Glutamine Mimic: SARS-CoV-2 3CL Protease Inhibition, Coronavirus Antiviral Activity, and in Vitro Biological Stability. J. Med. Chem. 2021.
- Ma, C.; Hu, Y.; Townsend, J.A.; Lagarias, P.I.; Marty, M.T.; Kolocouris, A.; Wang, J. Ebselen, Disulfiram, Carmofur, PX-12, Tideglusib, and Shikonin Are Nonspecific Promiscuous SARS-CoV-2 Main Protease Inhibitors. ACS Pharmacol. Transl. Sci. 2020, 3, 1265–1277.
- Ma, C.; Tan, H.; Choza, J.; Wang, Y.; Wang, J. Validation and invalidation of SARS-CoV-2 main protease inhibitors using the Flip-GFP and Protease-Glo luciferase assays. Acta Pharm. Sin. B 2021.
- Sun, L.-Y.; Chen, C.; Su, J.; Li, J.-Q.; Jiang, Z.; Gao, H.; Chigan, J.-Z.; Ding, H.-H.; Zhai, L.; Yang, K.-W. Ebsulfur and Ebselen as highly potent scaffolds for the development of potential SARS-CoV-2 antivirals. Bioorg. Chem. 2021, 112, 104889.
- Amporndanai, K.; Meng, X.; Shang, W.; Jin, Z.; Rogers, M.; Zhao, Y.; Rao, Z.; Liu, Z.-J.; Yang, H.; Zhang, L.; et al. Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives. Nat. Commun. 2021, 12, 3061.
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W.; et al. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Commun. 2021, 12, 3623.
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol. Transl. Sci. 2021, 4, 1096–1110.
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Li, S.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzyme Inhib. Med. Chem. 2021, 36, 497–503.
- Nguyen, T.T.H.; Jung, J.-H.; Kim, M.-K.; Lim, S.; Choi, J.-M.; Chung, B.; Kim, D.-W.; Kim, D. The Inhibitory Effects of Plant Derivate Polyphenols on the Main Protease of SARS Coronavirus 2 and Their Structure–Activity Relationship. Molecules 2021, 26, 1924.