Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Infectious Diseases
PLG0206, an engineered cationic antibiotic peptide that is 24 residues long, has been designed to address some limitations of other natural antimicrobial peptides (AMPs), such as toxicity, pharmacokinetics, and limited activity due to pH and ion concentrations.
- periprosthetic joint infections
- antimicrobial peptides
- antibiotic resistance
1. Introduction
Antibiotics have been used as successful treatments for many bacterial diseases, but the increasing prevalence of antibiotic-resistant microorganisms is a major public health concern. Antimicrobial peptides (AMPs) have gained much attention for their potential to treat diseases related to bacterial and viral infections, specifically against pathogens that have become multidrug resistant (MDR), extensively drug resistant (XDR), or pan-drug resistant (PDR) [1,2].
AMPs are a class of antimicrobial effector molecules of 10–50 amino acids in length that contain an amphipathic structure as a consensus motif and are found in many species across all kingdoms of life. As components of the mammalian innate immune system, they contribute to the first line of defense against invading pathogens [3,4]. Human cathelicidin (LL-37) broadly represents the class of AMPs and is the best characterized of the natural AMPs. LL-37 exhibits both antimicrobial activity and host immunoregulatory properties that are common among naturally occurring molecules [5]. However, AMPs have not demonstrated significant success therapeutically, primarily due to their toxicity when administered systemically and activity that is highly sensitive to pH and ion concentrations [3,6].
Engineered cationic antibiotic peptides (eCAPs) such as PLG0206—also known as WLBU2—have been designed to mitigate the shortcomings of natural AMPs and exhibit a broadened spectrum of activity, with increased activity in complex biological environments and an improved systemic safety profile [7]. PLG0206 is a 24-residue de novo eCAP [8]. PLG0206 has 13 arginine residues, 8 valine residues, and 3 tryptophan residues in the hydrophobic face that are separated from each other by at least seven amino acids (RRWVRRVRRWVRRVVRVVRRWVRR) [7]; the predicted structure is shown in Figure 1 [9,10,11].
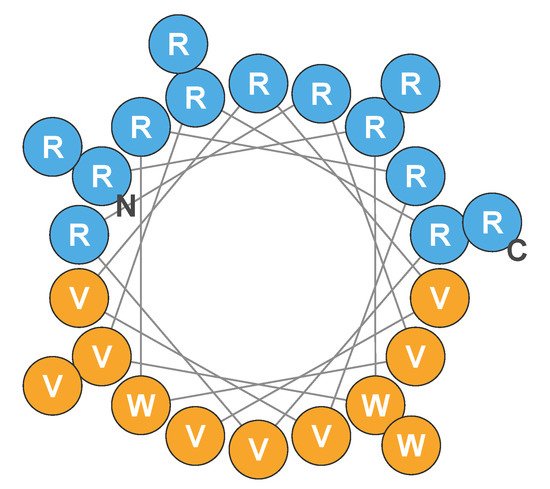
Figure 1. Helical wheel of the predicted structure of PLG0206. Blue circles indicate hydrophilic (cationic) amino acid residues; orange circles represent hydrophobic residues. C, carboxy terminus; N, amino terminus; R, arginine; V, valine; W, tryptophan. Adapted from Deslouches et al., 2005, 2015, and 2020 [9,10,11].
Due to its antibacterial and antibiofilm activity, PLG0206 may have the unique potential to successfully address infections related to implanted medical devices, which are prone to biofilm-related infections that are difficult to eradicate. One of the potential applications for PLG0206 is treating infections in humans that occur after joint replacement. Periprosthetic joint infections (PJIs) following total knee arthroplasty and other types of joint replacements can be a major problem for patients, caregivers, and health care providers; these infections are potentially fatal [12]. PJIs have an estimated annual incidence of 2% in both total hip and total knee replacements [13]. These infections can occur at different intervals—early, delayed, and late onset—depending on the type of pathogen, and it is believed that more virulent microorganisms are responsible for early-onset PJIs that occur within 3 months after surgery. Prominent bacteria that cause early infections include Staphylococcus aureus, Enterococcus spp., and aerobic Gram-negative bacilli, as well as coagulase-negative staphylococci [14,15]. Approximately 31–36% of early infections are polymicrobial and may require broad-spectrum antimicrobial therapy [14,16].
When PJIs are diagnosed in the acute phase, their treatment includes debridement of the wound (i.e., irrigation and debridement, I&D) and 6 weeks of intravenous antibiotic therapy [17]. I&D has a high long-term treatment failure rate of approximately 57% at 4 years [12]. In most cases, I&D failure is followed by a two-stage revision and removal of the prosthesis in 54.1% of cases [12]. All options after treatment failure require surgical procedures with rehospitalization, immobility, and compromised daily functioning, resulting in a 20% risk of mortality within 5 years after an I&D procedure or the initial surgical intervention [12,18]. A major cause of failure of I&D is the presence of a biofilm, leading to bacterial “persisters” that result in extreme difficulty in eradicating the pathogen from the implant [19,20]. Cefazolin, rifampin, nafcillin, ceftriaxone, and vancomycin are commonly recommended antibiotics used for a 6-week intravenous treatment after I&D [17], but these antibiotics are inadequate to remove S. aureus biofilm from prostheses [19]. There are currently no drugs specifically indicated to disrupt biofilms or to target the “persister” pathogens in biofilms.
2. Pharmacology
2.1. Preclinical Pharmacology
2.1.1. Mechanism of Action
Several studies have examined how PLG0206 exhibits bactericidal efficacy. Heinrich et al. [21] showed that neither the secondary structure nor pore formation is a critical determinant of bactericidal efficacy for PLG0206. The interaction of PLG0206 with the bacterial cell membrane leads to lipid phase consolidation, resulting in localized stiffening [22], ordering, and alteration of the thickness of the membranes [21]. Consequently, this disruption of the bacterial cell membrane results in lipid headgroup spacing mismatch and lowering of the energy barrier to ion flow across the membrane.
2.1.2. Selectivity for Bacterial Cells
The selectivity of PLG0206 for bacterial membranes is driven, in part, by the electrostatic interaction between the cationic charge of the peptide and the net negatively charged bacterial membranes. The potential toxicity of PLG0206 to eukaryotic cells, which also carry a net negative charge, was investigated by co-culturing Pseudomonas aeruginosa with primary human skin fibroblasts [9], peripheral blood mononuclear cells [7], and red blood cells [23] in the presence of PLG0206. At the end of the incubations, killing of P. aeruginosa versus lysis of the human cells was determined. These studies demonstrated that PLG0206 had little to no cytotoxic effects on the primary human cells at concentrations that effectively killed P. aeruginosa.
In preclinical studies, PLG0206 in phosphate-buffered saline (PBS) was not toxic to human red blood cells (up to 20 µM) and demonstrated low hemolytic potential (up to 10 µM) [23]. PLG0206 in whole human blood also demonstrated no toxicity and low hemolytic potential (to 50 µM) [7]. These data provide encouraging evidence that PLG0206 is not toxic to human blood cells and may be a viable systemic treatment in humans.
2.1.3. Degradation
The proteolytic activity associated with red blood cells is distinct from that of serum proteases and must be considered for any AMPs under development, as quicker degradation can result in loss of activity [24]. When incubated with cytosolic extracts of red blood cells, PLG0206 at 20 μM had a slow degradation profile and performed better than most other natural and synthetic peptides previously studied [24]. Furthermore, the robustness of intravenously administered PLG0206 to proteolytic degradation was demonstrated in a Phase 1 clinical study showing a median terminal half-life of up to 19.97 h [25].
2.1.4. Pharmacokinetics and Absorption, Distribution, Metabolism, and Excretion
The absorption, distribution, metabolism, and excretion (ADME) of PLG0206 have been initially characterized. In human plasma, the plasma protein binding level of PLG0206 was determined to be ~98%, which is similar to the levels observed in animal plasma. Administration of radiolabeled PLG0206 in mice [26] showed rapid distribution to kidneys and spleen. Although the metabolism of PLG0206 has not been definitively evaluated, all tissues examined contained measurable PLG0206-derived 14C. The majority of an injected 14C dose recovered in non-carcass tissues was detected in the liver, kidney, and plasma at 5 min post-dose (32.8%, 9.0%, and 7.75%, respectively), 15 min post-dose (36.3%, 11.1%, 5.98%, respectively), and 24 h post-dose (8.6%, 2.6%, and 1.5%) [27]. The average recovery of 14C in the urine was 6.29% between 0 and 16 h and 12.5% between 0 and 24 h. The average recovery of 14C in the feces was 0.73% between 0 and 16 h and 2.2% between 0 and 24 h. Thus, ~2.3% of the dose was excreted over 24 h. Of the total 14C dose injected, the mean recovery was 106.6%, 99.4%, and 65.5% at 5 min, 15 min, and 24 h post-dose, respectively [26,27].
The pharmacokinetics of PLG0206 were consistent across animal models. Exposure, in terms of area under the plasma concentration time curve from time 0–t (AUC[0–t]) and the maximum plasma concentration (Cmax), increased with increasing dose in a greater than dose-proportional manner. The total plasma clearance of PLG0206 was considered moderate to high in animals, with a tendency to decrease clearance with an increasing dose. The calculated terminal elimination half-life of PLG0206 following a single dose was approximately 2 h in rats and 8 h in cynomolgus monkeys [26].
In a rabbit femorotibial joint debridement model, PLG0206 at 0.4, 1.2, and 3.6 mg in 0.9% saline was applied to the joint at a volume of 2 mL/animal (dose concentrations of 0.2, 0.6, and 1.8 mg/mL) for 30 min (twice the planned 15-min duration in human studies). Blood samples were taken pre-dose and up to 24.5 h after the start of exposure (SOE). Plasma PLG0206 concentrations were below the lower limit of quantitation (LLOQ; 5 ng/mL) in plasma samples at all time points from animals in the 0.4- and 1.2-mg groups. In the 3.6-mg group, PLG0206 was quantifiable for up to 2 h post-SOE, and the time to maximum plasma concentration (Tmax) was observed at 0.533 h post-SOE. The Cmax was 9.30 ng/mL, and the AUC0–24.5 was 9.68 ng*h/mL. Additionally, in a local administration minipig femorotibial joint debridement model, the animals underwent surgery, and the femorotibial joint was irrigated with a vehicle or PLG0206 at doses of 3, 10, and 30 mg/mL (15, 50, or 150 mg within 5 mL/animal). The mean Cmax values for these doses were 2.30, 0.454, and 13.6 ng/mL, respectively [28]. Thus, systemic exposure was expected to be low in humans following irrigation with PLG0206.
A population pharmacokinetic model was developed from the observed PLG0206 pharmacokinetic data pooled from a total of nine animal studies (one in mice, three in rats, one in dogs, and four in monkeys). PLG0206’s pharmacokinetics were best described by a three-compartment zero-order absorption model. A population pharmacokinetic model showed that the bioavailability of PLG0206 increases with increasing weight (which is associated with the size of the species). This means that bioavailability is higher in larger species for the same milligram-per-kilogram dosage. Overall, the population pharmacokinetic model described the observed data in all species and was deemed appropriate for use in simulations of pharmacokinetic profiles in animals and extrapolation to humans using body weight (by allometric scaling). There was no effect of sex on pharmacokinetics observed in any of the species. Finally, based on the rabbit and minipig data presented above, it appears that systemic exposure following application of PLG0206 to surgically exposed femorotibial joint is minimal due to poor absorption from the space.
2.1.5. Preclinical Safety
A comprehensive battery of in vitro and in vivo safety pharmacology studies has been completed for PLG0206. In an in vitro study in human embryonic kidney cells and Chinese hamster ovary cells, PLG0206 (0.1 μM) had no biologically meaningful effect on KvLQT1/mink and little to no effect on either hERG or Nav1.5 channel currents. In a cardiovascular assessment study, PLG0206 administered in an intravenous infusion (1 h) at doses of 3 and 6 mg/kg via a single dose and 15 mg/kg for 10 consecutive days to conscious cynomolgus monkeys was not associated with any changes in the electrocardiogram (PR, QRS, QT, QTcF). No effects of PLG0206 were observed on the respiratory system of rats following a single intravenous infusion (1 h) at a dose of ≤6 mg/kg body weight [28]. Overall, the available pharmacology data for PLG0206, including the safety data from cardiovascular, respiratory, and central nervous system function studies, support the development of PLG0206 as a therapeutic for PJI, where systemic exposure is expected to be very low.
PLG0206 was evaluated for the inhibition of radioligand binding to a broad panel of clinically important receptors, ion channels, and transporters and for inhibition of the activity of a panel of enzymes. At a concentration of 10 μM, PLG0206 exhibited >50% binding to a handful of receptors, channels, transporters, and enzymes. The functional activity of PLG0206 on these targets is currently being evaluated in secondary functional assays.
Importantly, all genetic toxicology studies for PLG0206 were negative, indicating a low genotoxic potential. In the nonclinical studies, PLG0206 was formulated in 0.9% sodium chloride, which is identical to the formulated clinical drug product for the first-in-human study.
To evaluate systemic safety, PLG0206 was administered to rats at doses of 0, 1, 3, and 6 mg/kg via 1-h infusions once daily for 14 days. In this study, PLG0206 appeared to be safe and well tolerated. In a different model, PLG0206 was administered once daily at 3, 6, and 12 mg/kg via a 1-h infusion for 14 days in cynomolgus monkeys, and PLG0206 was well tolerated [28]. No safety issues were noted in the femorotibial joint debridement studies in minipigs and rabbits when PLG0206 was applied to the joint for 30 min. Overall, systemic administration and joint application of PLG0206 were well tolerated and safe [28].
2.2. Human Pharmacokinetics
The pharmacokinetics of PLG0206 were evaluated in 33 healthy volunteers, as summarized in Figure 2. PLG0206 was administered intravenously and exhibited linear pharmacokinetics over the dose range of 0.05–1.0 mg/kg. The median terminal elimination half-life was 7.37–19.97 h [25]. The AUC(0–t) ranged from 1283.74 h*ng/mL at 0.05 mg/kg to 12,612.56 h*ng/mL at 1 mg/kg. The AUC(0–∞) was between 1581.41 h*ng/mL at 0.05 mg/kg and 21,141.52 h*ng/mL at 1 mg/kg. The Cmax (mean ± SD) increased from 256 ± 58 ng/mL in Cohort 1 (0.05 mg/kg, intravenous, 1 h) to 2653 ± 719 ng/mL in Cohort 5 (1 mg/kg, intravenous, 4 h) [25]. The mean apparent volume of distribution increased from 25.49 L in Cohort 1 to 94.2 L in Cohort 5. The mean clearance values (range, 2.42–4.18 L/h) were similar across all PLG0206 doses and infusion times [25].
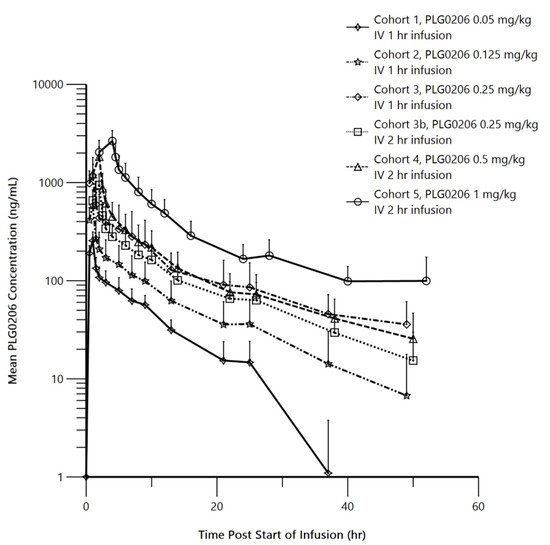
Figure 2. Mean (SD) PLG0206 plasma concentration (in log scale) time plots by dose group. IV, intravenous; hr, hour. Reproduced with permission from Huang et al., 2021 [25].
This entry is adapted from the peer-reviewed paper 10.3390/antibiotics11010041
This entry is offline, you can click here to edit this entry!