The main protease (M-pro) or 3C-like protease of coronaviruses plays an essential role in virus replication. This protease contributes to the cleavage of the ppa1a and pp1ab polyproteins to produce several non-structural proteins, including M-pro itself. Since the beginning of the COVID-19 pandemic, the SARS-CoV-2 M-pro enzyme has been extensively studied, and its inhibitors are promising effective drugs for fighting against SARS-CoV-2. The first attempts to discover SARS-CoV-2 M-pro inhibitors used previously developed protease inhibitors or tried to repurpose drugs from other diseases. Covalent inhibitors form a covalent bond, usually with catalytic Cys145. Non-covalent inhibitors bind by non-covalent interactions at the active site of the enzyme, inhibiting its function.
1. Introduction
Since the onset of the COVID-19 pandemic, the scientific community has focused on studying the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus that causes the disease and on developing therapies and vaccines, several of which have been developed in record time. In the pharmacological field, no drugs have yet been definitively approved to inhibit the replication of SARS-CoV-2 and stop the development of COVID-19. Several targets are being studied, including the main protease (M-pro), which plays an essential role in virus replication [
1]. This protease and the papain-like protease cleave the pp1a and pp1ab polyproteins to produce several nonstructural proteins, including M-pro itself, required for virus replication and transcription [
1]. The high conservation of M-pro among related viruses, the importance of M-pro in the replication of the virus and the fact that M-pro only exists in coronaviruses and not in humans makes it an attractive target for the development of antiviral drugs [
2,
3,
4]. SARS-CoV-2 M-pro has 306 amino acids that form three domains (I, II and III) [
4]. The M-pro binding site is located between domains I and II, and domain III is involved in dimerization, which is essential for M-pro activity [
4]. Similar to the M-pro from SARS-CoV-1 and other coronaviruses, SARS-CoV-2 M-pro has two catalytic amino acids, His41 and Cys145 (
Figure 1). A catalytic water molecule is also important and makes a strong hydrogen bond with His41 [
5]. Although some allosteric binding sites have been identified for the SARS-CoV-2 M-pro [
6,
7,
8,
9], most of the inhibitors crystallized within the M-pro bind to the active site [
10]. One strategy used to find SARS-CoV-2 M-pro inhibitors, especially at the beginning of the pandemic, was drug repositioning [
2,
11,
12,
13]. This strategy is based on looking for drugs approved for one disease (therefore, its safety and possible adverse effects are known) that can be used to treat another—in this case, COVID-19. One of the most widely used computational tools for repositioning drugs, or looking for compounds with new activities, is what is known as protein-ligand docking. This tool predicts whether a particular molecule can bind (and, if it can, how) to a particular target (for example, the SARS-CoV-2 M-pro [
14]). However, protein-ligand docking has several limitations, such as the consideration of the protein as a rigid body and the lack of confidence in the ability of scoring functions to give accurate binding energies [
15,
16]. In addition, the flexibility of the SARS-CoV-2 M-pro makes it a challenging target for small-molecule inhibitor design [
17]. Using two different SARS-CoV-2 M-pro structures and five protein-ligand docking methods, we have recently shown that docking scores or the Gibbs free energy (∆G) calculated with an MM-GBSA method [
18] do not correlate with bioactivity [
19], probably because of the inability of common docking programs to correctly reproduce the binding modes of SARS-CoV-2 M-pro inhibitors [
20]. This reinforces the idea that it is essential to validate the results obtained by protein-ligand docking or any other computational tool, especially when analyzing SARS-CoV-2 M-pro inhibitors [
19,
21,
22,
23]. The results of protein-ligand docking can be computationally validated by re-docking, cross-docking and applying the same protocol to a set of known active compounds and a set of decoy or inactive compounds [
19]. Protein-ligand docking is expected to discriminate decoys from active compounds. If docking scores are used to rank the potency of a set of compounds, it must first be demonstrated that there is a correlation between docking scores and activity or potency, for example, expressed as IC
50 values [
19]. However, the best way to validate the predictions of protein-ligand docking is to experimentally test the predicted bioactivity of selected hits.
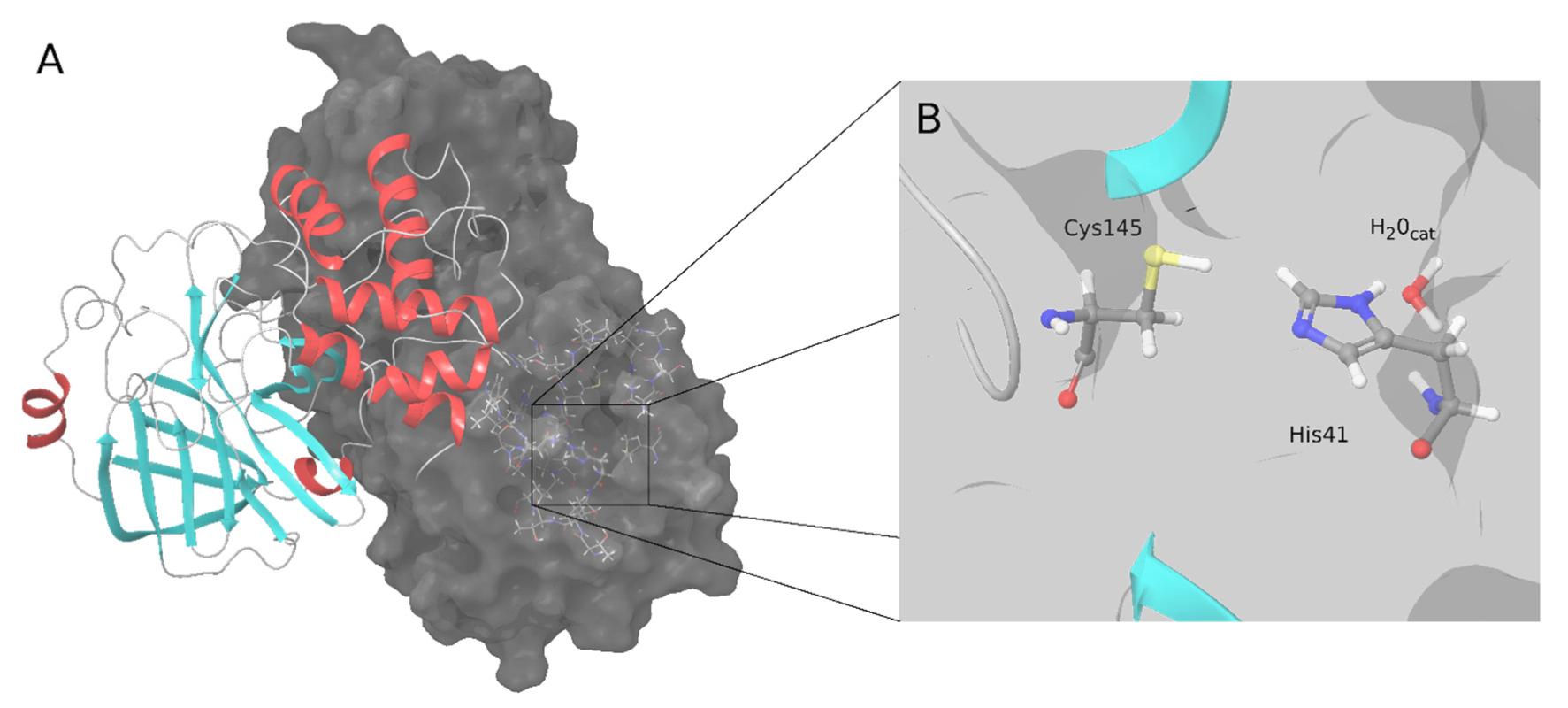
Figure 1. SARS-CoV-2 M-pro structure. (A) Biological assembly of the M-pro in its dimeric form. The left protomer is shown in cartoon representation, colored by protein secondary structure, and the right protomer is displayed as a surface. (B) Detailed snapshot of the catalytic water, Cys145 and His41.
Since the beginning of the COVID-19 pandemic, developing SARS-CoV-2 M-pro inhibitors has been an active area of research. However, it did not have to start from scratch. Previous research about protease inhibitors, especially from SARS-CoV and MERS-CoV, proved to be useful [
24,
25]. Known inhibitors of proteases from HIV and Hepatitis C virus, in addition to calpain and caspase-3 inhibitors, were systematically analyzed to test their capacity to inhibit the SARS-CoV-2 M-pro [
26]. Compounds developed against the M-pro of other coronaviruses were also tested, and some were found to be potent SARS-CoV-2 M-pro inhibitors [
24,
27,
28]. The complete genome sequence of SARS-CoV-2 [
29] and the first crystallized structure of the SARS-CoV-2 M-pro [
4] were two important milestones in the development of new SARS-CoV-2 M-pro inhibitors. The article describing the first crystallized SARS-CoV-2 M-pro structure (the 6LU7 structure) also presented the first SARS-CoV-2 M-pro inhibitors [
4]. These first inhibitors included the N3 compound, which had previously been developed as a protease inhibitor for multiple coronaviruses, including SARS-CoV and MERS-CoV, approved drugs (such as disulfiram and carmofur) and preclinical or clinical-trial drug candidates (ebselen, shikonin, tideglusib, PX-12 and TDZD-8) [
4]. Since then, thousands of compounds have been suggested as SARS-CoV-2 M-pro inhibitors through computational methods such as protein-ligand docking, high-throughput screening experiments, computer-aided design and synthesis of new compounds. Several articles have reviewed the SARS-CoV-2 M-pro inhibitors discovered to date [
25,
28,
30,
31,
32,
33,
34,
35,
36,
37,
38].
2. SARS-CoV-2 M-Pro Inhibitors
Table 1 shows the origin of the non-redundant set of 1765 SARS-CoV-2 M-pro inhibitors collected between January 2020 and August 2021 (see
supplementary file S1). This set of inhibitors includes only those compounds whose inhibitory capacity, mainly expressed as the IC
50 value, against M-pro from SARS-CoV-2 has been determined. A total of 758 compounds were extracted from peer-reviewed articles published between January 2020 and August 2021. When multiple IC
50 values were found for the same compound, the mean value was calculated. From a set of 1037 M-pro inhibitors with an IC
50 value downloaded from the COVID Moonshot project [
39,
40] on 1st October 2021, the compounds that had already been collected from the bibliographic search were discarded. In the end, 999 compounds were collected from COVID Moonshot. The IC
50 values of these compounds were estimated as the mean value of the IC
50 values from two biochemical assays: a fluorescence-based assay and a RapidFire Mass Spectrometry assay. Finally, 8 compounds were collected from the ChEMBL database [
41], which contained more SARS-CoV-2 M-pro inhibitors, but most of them had already been collected from the bibliography. The SMILES of the 1765 SARS-CoV-2 M-pro inhibitors were standardized with the Standardizer 21.15.0 program from ChemAxon (
http://www.chemaxon.com, accessed on 4 September 2021). The pIC
50 values of the SARS-CoV-2 M-pro inhibitors collected range from 2.5 to 9.0 (
Table 1). Putative covalent inhibitors were identified by the presence of typical covalent warheads (
Table 2). When one of these warheads is in the appropriate position within the M-pro binding site, it can form a covalent bond, usually with the catalytic Cys145 [
25]. There are twice as many non-covalent inhibitors as putative covalent inhibitors (
Table 1), although pIC
50 values are highest in some putative covalent inhibitors (
Table 1 and
Figure 2). However, conventional IC
50 measurements are of limited value for characterizing the potency of irreversible covalent inhibitors, because incubation for different periods of time would provide different IC
50 values [
42]. Other parameters, such as molecular weight, LogP, number of hydrogen bond donors and hydrogen bond acceptors were similar between the covalent and non-covalent sets.
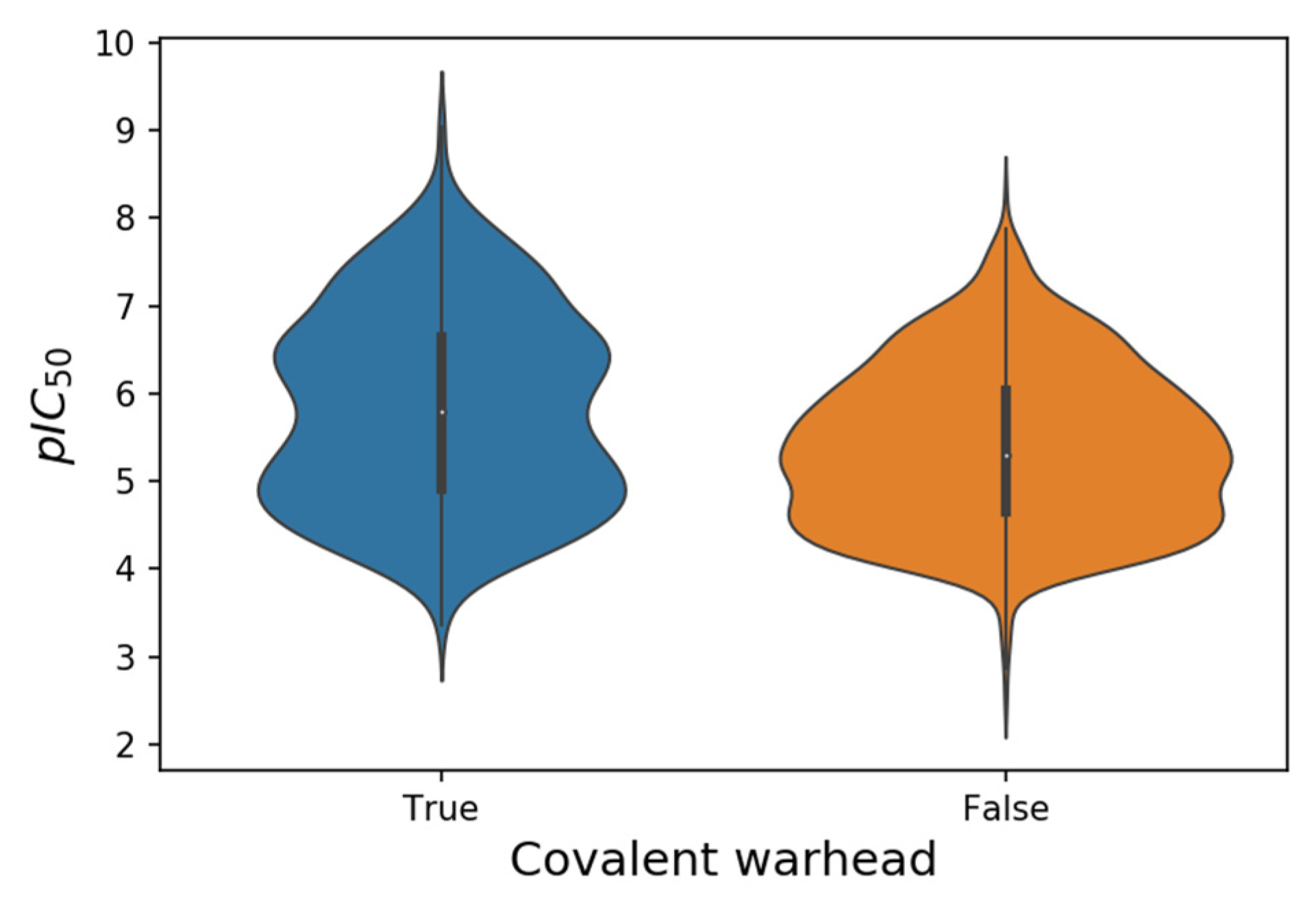
Figure 2. Violin plots of the pIC50 values from 552 putative covalent and 1213 non-covalent SARS-CoV-2 M-pro inhibitors.
Table 1. Number of SARS-CoV-2 M-pro inhibitors collected.
| SARS-CoV-2 M-Pro Inhibitor Set |
Number of Compounds (Covalent/Non-Covalent) 1 |
pIC50 Range |
pIC50 Range Covalent |
pIC50 Range Non-Covalent |
| From the bibliography |
758 (346/412) |
2.5–9.0 |
3.4–9.0 |
2.5–8.3 |
| From COVID Moonshot |
999 (205/794) |
4.0–7.8 |
4.0–7.8 |
4.0–7.4 |
| From ChEMBL |
8 (1/7) |
5.4–6.1 |
5.4 |
5.5–6.1 |
| All |
1765 (552/1213) |
2.5–9.0 |
3.4–9.0 |
2.5–8.3 |
Table 2. Covalent warheads that can be used to identify putative covalent inhibitors. It shows the SMARTS that can be used to identify each warhead and some examples of SARS-CoV-2 inhibitors that contain each warhead. These covalent warheads were used to identify putative covalent inhibitors among the known SARS-CoV-2 M-pro inhibitors.
| Warhead |
SMARTS |
Examples |
| Acrylamide |
[C;H2:1]=[C;H1]C(N)=O |
CVD-0004255 |
| Chloroacetamide |
Cl[C;H2:1]C(N)=O |
BFC204 |
| Vinylsulfonamide |
NS(=O)([C;H1]=[C;H2:1])=O |
|
| Nitrile |
N#[C:1]-[*] |
Isavuconazole |
| Michael acceptors |
C=!@CC=[O,S] |
Cinanserin, MPI2, MPI9, N3 |
| Alpha-ketoamide |
C(=O)(C=O)N |
Boceprevir, narlaprevir, telaprevir, UAWJ248 |
| Aldehyde |
[CX3H1](=O) |
GC373, MI-05, MI-06, MI-09, MI-11, MI-13, MI-14, MI-21, MI-23, MI-28 |
| Bisulfite adduct of aldehyde |
C(O)S(=[OX1])([O])(=[OX1]) |
GC376 |
| Urea carbonyl |
[NX3][CX3](=[OX1])([NX3,nX3]) |
Carmofur |
| Bis(dialkylaminethiocarbonyl)disulfide |
[CX3](=[SX1])SS[CX3](=[SX1]) |
Disulfiram |
| Carbamoylsulfanyl |
[NX3,nX3][C,c](=[OX1])([SX2,sx2]) |
Tideglusib |
| Disulfide |
[SX2][SX2] |
PX-12 |
| Hydroxymethyl ketone |
[CX3H0](=[OX1])[CH2][OH] |
PF-00835231 |
| Alkoxymethyl ketone |
[CX3H0](=[OX1])[CH2][OX2H0] |
2683066-41-1, 2683066-42-2, 2683066-47-7 |
| Acyloxymethyl ketone |
[CX3H0](=[OX1])[CH2][OX2H0][CX3H0](=O) |
2683066-41-1, 2683066-42-2, 2683066-47-7 |
| Fluoro, Chloro-methyl ketone |
[CX3H0](=[OX1])[CH2][Cl,F] |
Z-AVLD-FMK |
| Ebselen related |
[Se]n(c=O) |
Ebselen |
Figure 3 shows the t-Distributed Stochastic Neighbor Embedding (t-SNE) visualization of the chemical space of the set of SARS-CoV-2 M-pro inhibitors extracted from the bibliography. In this representation, more similar compounds are closer together. Peptidomimetic compounds, such as alpha-acyloxymethylketones, telaprevir, boceprevir, GC373 and their derivatives, which mimic natural peptide substrates, are closer together at the top left of the figure. Other clusters of compounds represent derivative compounds that have been synthesized from a lead compound to increase its bioactivity. Thus, derivatives from perampanel, ML300, ML188, ebsulfur, ebselen and myricetin form well-defined clusters. Perampanel derivatives are an example of a very successful increase in activity. Perampanel was first predicted as a SARS-CoV-2 M-pro inhibitor by consensus docking [
2]. This prediction was confirmed by Jorgensen and coworkers, although perampanel showed only an approximate IC
50 of 100–250 μM [
43]. The same authors also optimized this compound and synthesized several derivative compounds [
44,
45,
46]. Some of these perampanel derivatives have IC
50 values in the low nanomolar range and are some of the most potent non-covalent SARS-CoV-2 M-pro inhibitors found to date.
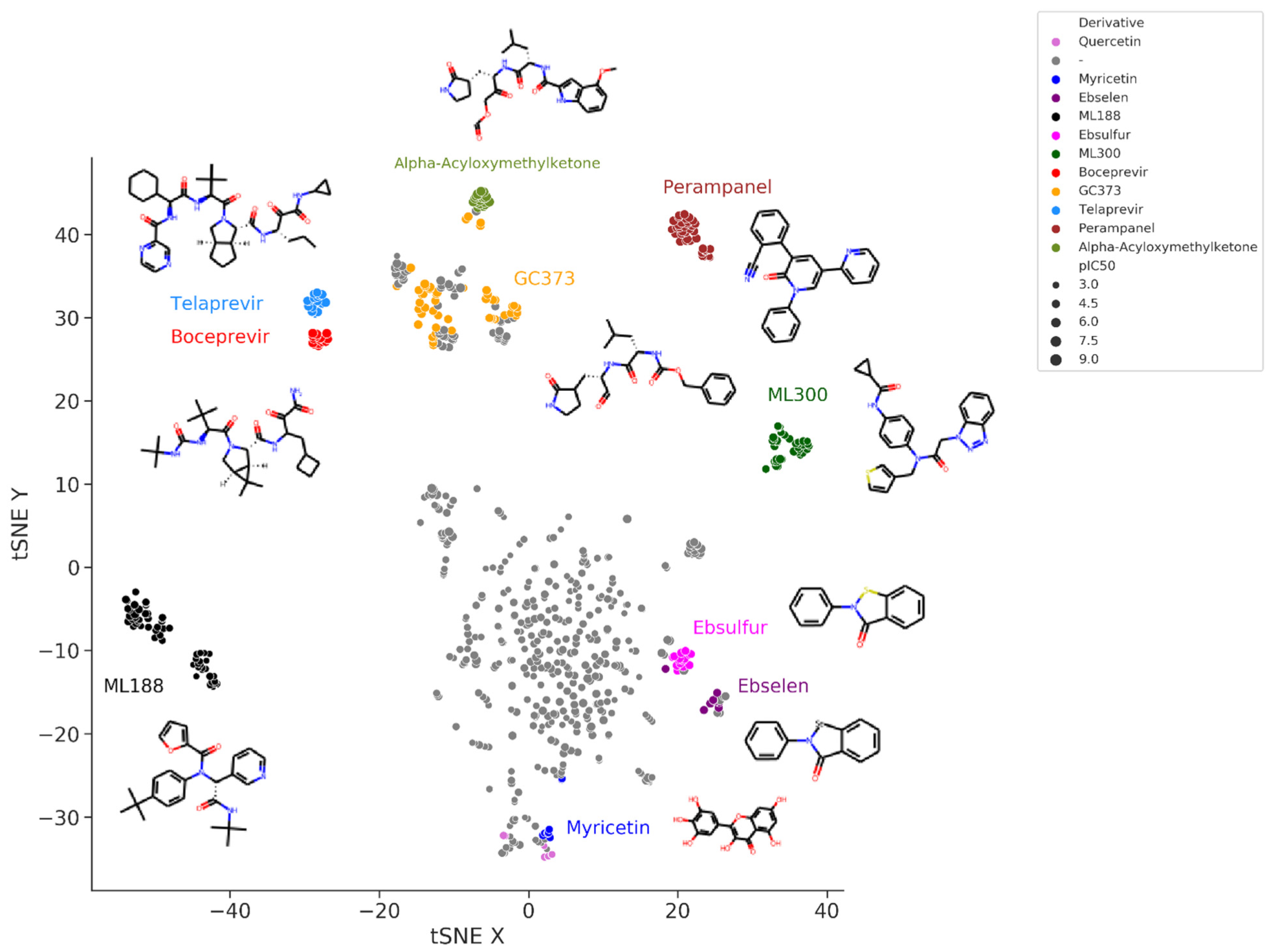
Figure 3. t-Distributed Stochastic Neighbor Embedding (t-SNE) visualization of the chemical space of a set of SARS-CoV-2 M-pro inhibitors extracted from the bibliography. Embedding is based on the 2048-bit Morgan fingerprint. Markers are colored according to several manually attributed chemotypes.
ML300 and ML188 are non-covalent inhibitors that were developed against the M-pro from SARS-CoV-1 [
47,
48]. Both compounds have been used to obtain more potent SARS-CoV-2 M-pro inhibitors that can inhibit SARS-CoV-2 replication in infected cells [
49,
50]. Boceprevir and telaprevir are approved protease inhibitors for treating hepatitis caused by the hepatitis C virus. Both compounds have been identified several times as covalent inhibitors of the SARS-CoV-2 M-pro [
43,
51,
52,
53,
54,
55,
56,
57,
58]. New bicycloproline derivatives have been designed and synthesized from them both [
59]. All compounds inhibited SARS-CoV-2 M-pro in vitro, with IC
50 values ranging from 7.6 to 748.5 nM [
59]. In addition, two of them, MI-09 and MI-30, showed excellent antiviral activity in a cell-based assay and significantly reduced lung viral loads and lesions in a transgenic mouse model of SARS-CoV-2 infection [
59]. GC376 is a covalent M-pro inhibitor that was developed as an inhibitor of the main protease of the feline coronavirus FCoV [
60] that also showed activity against the M-pro from MERS and SARS-CoV viruses [
61]. Its IC
50 activity against SARS-CoV-2 M-pro ranges between 0.026 and 0.89 μM [
51,
52,
61,
62,
63,
64,
65,
66,
67]. GC376 is a prodrug, and its bisulphite adduct is converted to an aldehyde to form GC373. This aldehyde forms a covalent interaction with the catalytic Cys145 of the SARS-CoV-2 M-pro [
61]. Several GC373 and GC376 derivative compounds have been designed and assayed [
63,
68,
69]. Some of them, such as UAWJ248 [
70], are more potent than GC376. A group of peptidomimetic compounds with an alpha-acyloxymethyl ketone warhead designed to form an irreversible covalent bond with Cys145 showed IC
50 values against the SARS-CoV-2 M-pro in the nM range [
71]. They also inhibited SARS-CoV-2 replication and presented low cytotoxicity and good stability [
71]. Ebselen is a covalent inhibitor of the SARS-CoV-2 M-pro, although its specificity has been questioned [
72,
73]. Several derivative compounds of ebselen and its sulfur derivative ebsulfur have been analyzed [
74,
75]. Some of the derivative compounds displayed more potent M-pro inhibition than ebselen and ebsulfur [
74,
75]. However, the promiscuous behavior of ebselen and ebsulfur and their lack of cellular antiviral activity [
74,
75] may also be applied to their derivatives. Myricetin is a flavonoid that acts as a non-peptidomimetic and covalent inhibitor of SARS-CoV-2 [
76,
77]. Its covalent behavior was unexpected and caused by the pyrogallol moiety that formed a covalent bond with Cys145 [
76]. Myricetin and its derivatives inhibit SARS-CoV-2 M-pro and SARS-CoV-2 replication in cells [
76,
77,
78,
79], and form a cluster at the bottom of
Figure 3, near quercetin and other flavonoids.
Several in vitro and in cellulo (using live cells) methods have been developed to measure the inhibitory potency of a compound against the SARS-CoV-2 M-pro. In vitro methods need to express and purify SARS-CoV-2 M-pro, so some tags are sometimes added. However, especially if they are located at the N-terminus, these tags can interfere with the binding of M-pro to its ligands. The activity values obtained by different laboratories or with different methods or conditions must be compared with great care. The presence of DTT has been reported to affect the inhibitory activity of covalent M-pro inhibitors. If the inhibitory effect of an M-pro inhibitor is eliminated or greatly reduced by the presence of DTT, the inhibition is not specific. Therefore, the potency of inhibition measured in the absence of DTT should not be used by itself. The potency of a compound to inhibit SARS-CoV-2 replication in cells cannot always be inferred from the potency to inhibit M-pro, determined in vitro. An antiviral assay that uses cells infected with SARS-CoV-2 provides a better estimate of the potency of a compound to inhibit virus replication. However, if it is to be ruled out that the toxicity of the compounds is responsible for the antiviral activity, the cytotoxicity of the compounds needs to be determined.
3. Conclusions
Although we have not yet hit the bullseye and no drug has yet been approved to inhibit SARS-CoV-2 M-pro, we may be close. Improving derivatives of a leading compound has proven to be a very successful strategy for finding potent SARS-CoV-2 M-pro inhibitors. Some derivative compounds designed in less than two years since the start of the COVID-19 pandemic represent an important step toward the development of new anti–SARS-CoV-2 drugs. Currently, there are several compounds with low nanomolar IC50 values against SARS-CoV-2 M-pro and high anti-SARS-CoV-2 efficacy in cell models, with values comparable to those of the FDA-approved RNA polymerase inhibitor remdesivir.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23010259