G protein-coupled receptors (GPCRs) represent the largest membrane protein family and a significant target class for therapeutics. Receptors from GPCRs’ largest class, class A, influence virtually every aspect of human physiology. About 45% of the members of this family endogenously bind flexible peptides or peptides segments within larger protein ligands. While many of these peptides have been structurally characterized in their solution state, the few studies of peptides in their receptor-bound state suggest that these peptides interact with a shared set of residues and undergo significant conformational changes.
- peptide GPCR
- class A GPCR
- peptide docking
- non-canonical amino acids
1. Introduction
1.1. G Protein-Coupled Receptors Are a Significant Target of Therapeutic Intervention
1.2. Peptide-Activated Receptors Are a Large Percentage of the GPCR Class A
1.3. Diversity of Peptide Ligands
| Peptide Modification | Example | Function |
|---|---|---|
| C-terminal amidation | Neuropeptide Y (NPY), neuromedin B | C-terminal amidation reduces the overall charge of a peptide, forms key hydrogen interactions that are important for the potency of the peptide [15], and increases the metabolic stability of peptides as well as their ability to resist enzymatic degradation [16] |
| N-terminal pyroglutamic acid | Thyroid stimulating hormone (TSH), gonadotropin-releasing hormone I (GnRHI), regulated upon activation, Normal T cell expressed, and presumably secreted (RANTES)/chemokine ligand 5 (CCL5) | The pyroglutamic acid is often involved in peptide-receptor recognition and potency [17], and provides stability against N-terminal degradation [18] |
| Bromination | Neuropeptides B and W (NPBW) | Bromination on N-terminal tryptophan might protect the peptide from amino-peptidases’ degradation [19] |
| Lipidation | Ghrelin | The attached lipid group (e.g., octanoyl group) is essential to the activity of the peptide [19] and affects the hydrophobicity of the peptide [20] |
| Disulfide bridge formation | Endothelin, vasopressin | The disulfide bonds stabilize the defined secondary structure [21], stabilizing the bound conformation of the peptide [22] |
| Differential proteolysis | Bradykinin, angiotensin, NPY/NPY3-36, apelin (Ape)-13/Ape-17/Ape 36, adrenocorticotropic hormone (ACTH), pro-opiomelanocortin (POMC) cleavage yielding α-, β-, and γ- melanocyte-stimulating hormone (MSH), and endorphins | Proteolysis can switch the activity of the peptides on and off [23] or differentiates the binding selectivity and the biological responses of the peptides [24] |
1.4. Reducing the Flexibility of Peptide Ligands Is Crucial for Success in Co-Crystallization
1.5. Complexity of Peptide Ligand and Receptor Interactions
2. Comparison of Peptide Binding Modes across Class A GPCRs
2.1. Diversity in the Binding Modes of the Peptide Ligands to Class A GPCRs
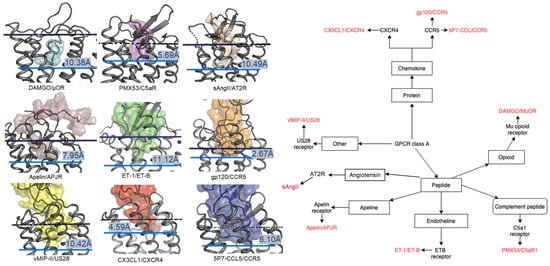
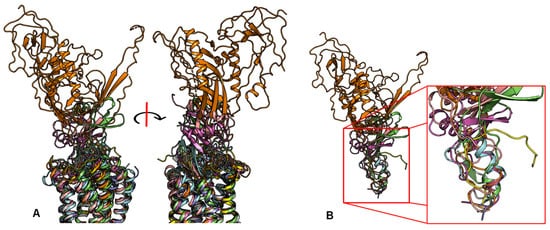
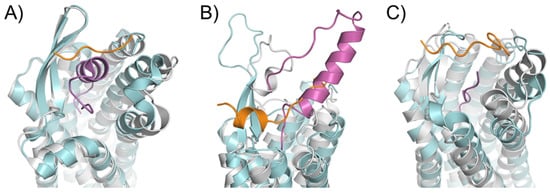
2.2. Peptide Ligands Affect the Conformation of the Extracellular Surface
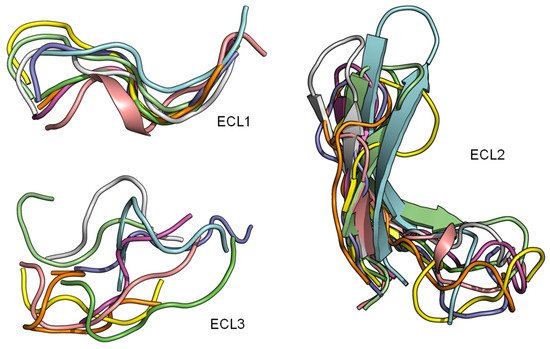
2.3. ECL1 and ECL2 Bound Conformation Have Conversed across Class A Peptide-GPCRs
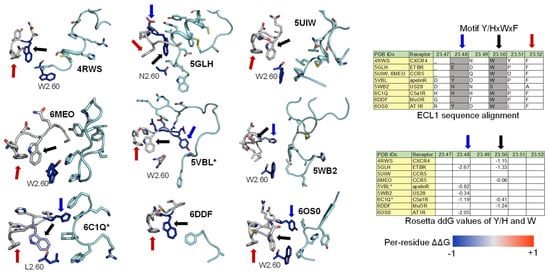
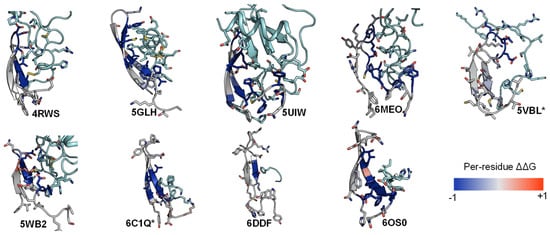
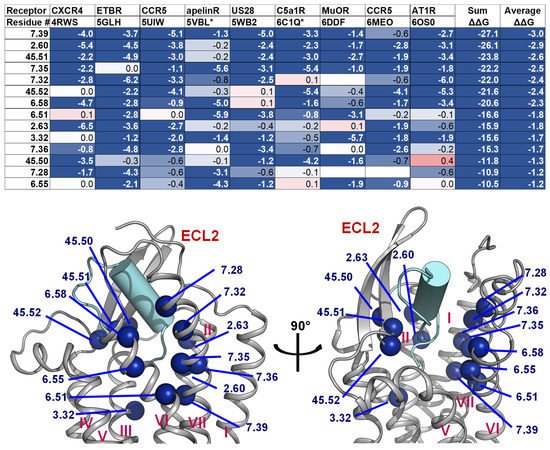
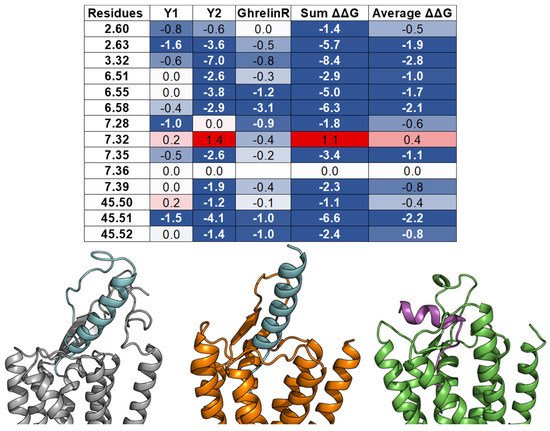
2.4. A List of 14 Common Interacting Residues Suggests a General Peptide Recognition and Binding Mechanism among Nine Class A GPCRs
This entry is adapted from the peer-reviewed paper 10.3390/molecules27010210
References
- Flock, T.; Hauser, A.S.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR-G-protein binding. Nature 2017, 545, 317–322.
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842.
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34.
- Isberg, V.; Mordalski, S.; Munk, C.; Rataj, K.; Harpsoe, K.; Hauser, A.S.; Vroling, B.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: An information system for G protein-coupled receptors. Nucleic Acids Res. 2017, 45, 2936.
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427.
- Bockaert, J.; Pin, J.P. Molecular tinkering of G protein-coupled receptors: An evolutionary success. EMBO J. 1999, 18, 1723–1729.
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272.
- Scott, L.J.; McCormack, P.L. Olmesartan Medoxomil. Drugs 2008, 68, 1239–1272.
- Markham, A.; Goa, K.L. Valsartan. Drugs 1997, 54, 299–311.
- Brogden, R.N.; Buckley, M.M.T.; Ward, A. Buserelin. Drugs 1990, 39, 399–437.
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 2020, 19, 389–413.
- Beck, A.; Bussat, M.C.; Klinguer-Hamour, C.; Goetsch, L.; Aubry, J.P.; Champion, T.; Julien, E.; Haeuw, J.F.; Bonnefoy, J.Y.; Corvaia, N. Stability and CTL activity of N-terminal glutamic acid containing peptides. J. Pept. Res. 2001, 57, 528–538.
- Chance, R.E. Pancreatic Polypeptide. In Encyclopedia of Hormones; Henry, H.L., Norman, A.W., Eds.; Academic Press: New York, NY, USA, 2003; pp. 142–146.
- Kaiser, A.; Muller, P.; Zellmann, T.; Scheidt, H.A.; Thomas, L.; Bosse, M.; Meier, R.; Meiler, J.; Huster, D.; Beck-Sickinger, A.G.; et al. Unwinding of the C-Terminal Residues of Neuropeptide Y is critical for Y2 Receptor Binding and Activation. Angew. Chem. Int. Ed. Engl. 2015, 54, 7446–7449.
- Xu, B.; Vasile, S.; Østergaard, S.; Paulsson, J.F.; Pruner, J.; Åqvist, J.; Wulff, B.S.; Gutiérrez-de-Terán, H.; Larhammar, D. Elucidation of the Binding Mode of the Carboxyterminal Region of Peptide YY to the Human Y2 Receptor. Mol. Pharmacol. 2018, 93, 323–334.
- Bradbury, A.F.; Smyth, D.G. Peptide amidation. Trends Biochem. Sci. 1991, 16, 112–115.
- Goren, H.J.; Bauce, L.G.; Vale, W. Forces and structural limitations of binding of thyrotrophin-releasing factor to the thyrotrophin-releasing receptor: The pyroglutamic acid moiety. Mol. Pharmacol. 1977, 13, 606–614.
- Morty, R.E.; Bulau, P.; Pellé, R.; Wilk, S.; Abe, K. Pyroglutamyl peptidase type I from Trypanosoma brucei: A new virulence factor from African trypanosomes that de-blocks regulatory peptides in the plasma of infected hosts. Biochem. J. 2006, 394, 635–645.
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660.
- Yang, J.; Brown, M.S.; Liang, G.; Grishin, N.V.; Goldstein, J.L. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 2008, 132, 387–396.
- Hewage, C.M.; Jiang, L.; Parkinson, J.A.; Ramage, R.; Sadler, I.H. Solution structure of a novel ETB receptor selective agonist ET1-21 by nuclear magnetic resonance spectroscopy and molecular modelling. J. Pept. Res. 1999, 53, 223–233.
- Hirata, Y.; Yoshimi, H.; Emori, T.; Shichiri, M.; Marumo, F.; Watanabe, T.X.; Kumagaye, S.; Nakajima, K.; Kimura, T.; Sakakibara, S. Receptor binding activity and cytosolic free calcium response by synthetic endothelin analogs in cultured rat vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1989, 160, 228–234.
- Hook, V.; Funkelstein, L.; Lu, D.; Bark, S.; Wegrzyn, J.; Hwang, S.-R. Proteases for processing proneuropeptides into peptide neurotransmitters and hormones. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 393–423.
- Wagner, L.; Wolf, R.; Zeitschel, U.; Rossner, S.; Petersén, Å.; Leavitt, B.R.; Kästner, F.; Rothermundt, M.; Gärtner, U.-T.; Gündel, D.; et al. Proteolytic degradation of neuropeptide Y (NPY) from head to toe: Identification of novel NPY-cleaving peptidases and potential drug interactions in CNS and Periphery. J. Neurochem. 2015, 135, 1019–1037.
- White, J.F.; Noinaj, N.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; Tate, C.G.; et al. Structure of the agonist-bound neurotensin receptor. Nature 2012, 490, 508–513.
- Da Costa, G.; Bondon, A.; Coutant, J.; Curmi, P.; Monti, J.P. Intermolecular interactions between the neurotensin and the third extracellular loop of human neurotensin 1 receptor. J. Biomol. Struct. Dyn. 2013, 31, 1381–1392.
- Ma, Y.; Yue, Y.; Ma, Y.; Zhang, Q.; Zhou, Q.; Song, Y.; Shen, Y.; Li, X.; Ma, X.; Li, C.; et al. Structural Basis for Apelin Control of the Human Apelin Receptor. Structure 2017, 25, 858–866.
- Burg, J.S.; Ingram, J.R.; Venkatakrishnan, A.J.; Jude, K.M.; Dukkipati, A.; Feinberg, E.N.; Angelini, A.; Waghray, D.; Dror, R.O.; Ploegh, H.L.; et al. Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 2015, 347, 1113–1117.
- Qin, L.; Kufareva, I.; Holden, L.G.; Wang, C.; Zheng, Y.; Zhao, C.; Fenalti, G.; Wu, H.; Han, G.W.; Cherezov, V.; et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 2015, 347, 1117–1122.
- Zheng, Y.; Han, G.W.; Abagyan, R.; Wu, B.; Stevens, R.C.; Cherezov, V.; Kufareva, I.; Handel, T.M. Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 2017, 46, 1005–1017.e5.
- Pedragosa-Badia, X.; Stichel, J.; Beck-Sickinger, A.G. Neuropeptide Y receptors: How to get subtype selectivity. Front. Endocrinol. (Lausanne) 2013, 4, 5.
- Joedicke, L.; Mao, J.; Kuenze, G.; Reinhart, C.; Kalavacherla, T.; Jonker, H.R.A.; Richter, C.; Schwalbe, H.; Meiler, J.; Preu, J.; et al. The molecular basis of subtype selectivity of human kinin G-protein-coupled receptors. Nat. Chem. Biol. 2018, 14, 284–290.
- Eisen, H.N.; Hou, X.H.; Shen, C.; Wang, K.; Tanguturi, V.K.; Smith, C.; Kozyrytska, K.; Nambiar, L.; McKinley, C.A.; Chen, J.; et al. Promiscuous binding of extracellular peptides to cell surface class I MHC protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4580–4585.
- Ngo, T.; Ilatovskiy, A.V.; Stewart, A.G.; Coleman, J.L.; McRobb, F.M.; Riek, R.P.; Graham, R.M.; Abagyan, R.; Kufareva, I.; Smith, N.J. Orphan receptor ligand discovery by pickpocketing pharmacological neighbors. Nat. Chem. Biol. 2017, 13, 235–242.
- Michino, M.; Beuming, T.; Donthamsetti, P.; Newman, A.H.; Javitch, J.A.; Shi, L. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol. Rev. 2015, 67, 198–213.
- Kooistra, A.J.; Kuhne, S.; de Esch, I.J.; Leurs, R.; de Graaf, C. A structural chemogenomics analysis of aminergic GPCRs: Lessons for histamine receptor ligand design. Br. J. Pharmacol. 2013, 170, 101–126.
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071.
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265.
- Chien, E.Y.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095.
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217.
- Wu, F.; Song, G.; de Graaf, C.; Stevens, R.C. Structure and Function of Peptide-Binding G Protein-Coupled Receptors. J. Mol. Biol. 2017, 429, 2726–2745.
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the delta-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404.
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332.
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326.
- Thompson, A.A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H.; Vardy, E.; Huang, X.P.; Trapella, C.; Guerrini, R.; Calo, G.; et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 2012, 485, 395–399.
- Zhang, C.; Srinivasan, Y.; Arlow, D.H.; Fung, J.J.; Palmer, D.; Zheng, Y.; Green, H.F.; Pandey, A.; Dror, R.O.; Shaw, D.E.; et al. High-resolution crystal structure of human protease-activated receptor 1. Nature 2012, 492, 387–392.
- Zheng, Y.; Qin, L.; Zacarias, N.V.; de Vries, H.; Han, G.W.; Gustavsson, M.; Dabros, M.; Zhao, C.; Cherney, R.J.; Carter, P.; et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 2016, 540, 458–461.
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390.
- Oswald, C.; Rappas, M.; Kean, J.; Dore, A.S.; Errey, J.C.; Bennett, K.; Deflorian, F.; Christopher, J.A.; Jazayeri, A.; Mason, J.S.; et al. Intracellular allosteric antagonism of the CCR9 receptor. Nature 2016, 540, 462–465.
- Yin, J.; Babaoglu, K.; Brautigam, C.A.; Clark, L.; Shao, Z.; Scheuermann, T.H.; Harrell, C.M.; Gotter, A.L.; Roecker, A.J.; Winrow, C.J.; et al. Structure and ligand-binding mechanism of the human OX1 and OX2 orexin receptors. Nat. Struct. Mol. Biol. 2016, 23, 293–299.
- Yin, J.; Mobarec, J.C.; Kolb, P.; Rosenbaum, D.M. Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature 2015, 519, 247–250.
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem. 2015, 290, 29127–29139.
- Asada, H.; Horita, S.; Hirata, K.; Shiroishi, M.; Shiimura, Y.; Iwanari, H.; Hamakubo, T.; Shimamura, T.; Nomura, N.; Kusano-Arai, O.; et al. Crystal structure of the human angiotensin II type 2 receptor bound to an angiotensin II analog. Nat. Struct. Mol. Biol. 2018, 25, 570–576.
- Cheng, R.K.Y.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.G.; Brown, G.A.; Cooke, R.M.; Dumelin, C.E.; Dore, A.S.; et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 2017, 545, 112–115.
- Shihoya, W.; Nishizawa, T.; Okuta, A.; Tani, K.; Dohmae, N.; Fujiyoshi, Y.; Nureki, O.; Doi, T. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature 2016, 537, 363–368.
- Yang, Z.; Han, S.; Keller, M.; Kaiser, A.; Bender, B.J.; Bosse, M.; Burkert, K.; Kogler, L.M.; Wifling, D.; Bernhardt, G.; et al. Structural basis of ligand binding modes at the neuropeptide Y Y1 receptor. Nature 2018, 556, 520–524.
- Yin, J.; Chapman, K.; Clark, L.D.; Shao, Z.; Borek, D.; Xu, Q.; Wang, J.; Rosenbaum, D.M. Crystal structure of the human NK1 tachykinin receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 13264–13269.
- Robertson, N.; Rappas, M.; Dore, A.S.; Brown, J.; Bottegoni, G.; Koglin, M.; Cansfield, J.; Jazayeri, A.; Cooke, R.M.; Marshall, F.H. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 2018, 553, 111–114.
- Tanaka, H.; Yoshida, T.; Miyamoto, N.; Motoike, T.; Kurosu, H.; Shibata, K.; Yamanaka, A.; Williams, S.C.; Richardson, J.A.; Tsujino, N.; et al. Characterization of a family of endogenous neuropeptide ligands for the G protein-coupled receptors GPR7 and GPR8. Proc. Natl. Acad. Sci. USA 2003, 100, 6251–6256.
- Shaik, M.M.; Peng, H.; Lu, J.; Rits-Volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2019, 565, 318–323.
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the micro-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552.
- Liu, H.; Kim, H.R.; Deepak, R.; Wang, L.; Chung, K.Y.; Fan, H.; Wei, Z.; Zhang, C. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 2018, 25, 472–481.
- Krumm, B.E.; Grisshammer, R. Peptide ligand recognition by G protein-coupled receptors. Front. Pharmacol. 2015, 6, 48.
- Tikhonova, I.G.; Gigoux, V.; Fourmy, D. Understanding peptide binding in Class A G protein-coupled receptors. Mol. Pharmacol. 2019, 96, 550–561.
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376.
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161, 833–844.
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048.
- Woolley, M.J.; Conner, A.C. Understanding the common themes and diverse roles of the second extracellular loop (ECL2) of the GPCR super-family. Mol. Cell. Endocrinol. 2017, 449, 3–11.
- Isberg, V.; de Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.-P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers-aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31.
- Conway, P.; Tyka, M.D.; DiMaio, F.; Konerding, D.E.; Baker, D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014, 23, 47–55.
- Smith, C.A.; Kortemme, T. Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J. Mol. Biol. 2008, 380, 742–756.
- Bender, B.J.; Vortmeier, G.; Ernicke, S.; Bosse, M.; Kaiser, A.; Els-Heindl, S.; Krug, U.; Beck-Sickinger, A.; Meiler, J.; Huster, D. Structural Model of Ghrelin Bound to its G Protein-Coupled Receptor. Structure 2019, 27, 537–544.
- Raveh, B.; London, N.; Zimmerman, L.; Schueler-Furman, O. Rosetta FlexPepDock ab-initio: Simultaneous folding, docking and refinement of peptides onto their receptors. PLoS ONE 2011, 6, e18934.
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814.
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 20–41.