Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Endocrinology & Metabolism
Lipotoxicity and glucotoxicity emerged as established mechanisms participating in the pathophysiology of obesity-related type 2 diabetes in general, and in the loss of β-cell function in particular. Glycerolipid/free fatty acid cycle as a protective pathway mediating active storage and recruitment of lipids.
- glucotoxicity
- fatty acids
- diabetes
- beta-cell
- pancreatic islets
- insulin secretion
- lipotoxicity
- obesity
1. Functioning of the Glycerolipid/NEFA Cycle
Glucose is the main coordinator of β-cell function, and its metabolism governs the metabolic fate of fatty acids. Upon glucose deprivation, cytosolic fatty acids activated in the form of Lc-CoA can be funneled into mitochondria toward β-oxidation to ensure sufficient energy supply supporting basal cell functions (Figure 1). A rise in blood glucose levels increases glucose-derived pyruvate and the activity of the TCA cycle independently of the energy requirement when it comes to the β-cell. This results in the production of citrate that escapes the mitochondria and activates the acetyl-CoA carboxylase (ACC) to generate cytosolic malonyl-CoA. In turn, malonyl-CoA inhibits the carnitine palmitoyltransferase I (CPT1), blocking the entry of Lc-CoA into mitochondria and disabling β-oxidation [1][2]. In this context, Lc-CoA are redirected towards other pathways, notably to the glycerolipid/NEFA cycle (Figure 1). Originally, this cycle was described in the adipocytes as a mechanism to promote lipid accumulation under the form of triacylglycerol (TAG or triglycerides) and was thought to be marginal in other cell types, in particular β-cells. However, with the deeper understanding of the mechanisms underlying GSIS and its amplifying pathway, the glycerolipid/NEFA (GL/NEFA) cycle appeared as a candidate for a source of metabolic intermediaries involved in the potentiation of insulin secretion.
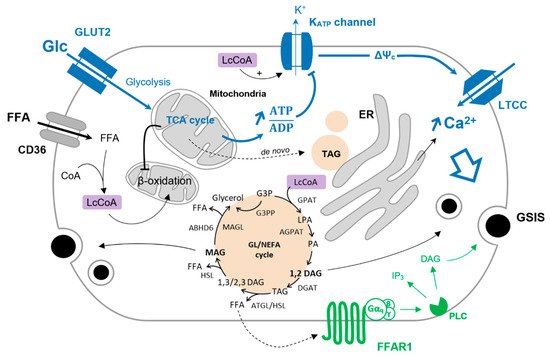
Figure 1. Glucose-induced FFA partitioning in the β-cell. Inhibition of β-oxidation by glucose (Glc) redirect Lc-CoA to the GL/NEFA cycle. Condensation of glycerol-trisphosphate and Lc-CoA is mediated by the glycerol-3-phosphate O-acyltransferase (GPAT) to form lysophosphatidic acid (LPA). LPA is further acetylated into phosphatidic by the 1-acyl-sn-glycerol-3-phosphate acyltransferase (AGPAT). The phosphohydrolase activity of lipins on PA produces 1,2-DAG that is then converted to TAG by the diacylglycerol O-acyltransferase (DGAT). TAG is sequentially hydrolyzed to 1/2,3-DAG, MAG, and finally glycerol by the adipose triglyceride lipase (ATGL), the hormone-sensitive lipase (HSL), and the MAG lipase (MAGL). MAG can also be hydrolyzed to glycerol by the α/β -hydrolase domain-containing 6 lipase (ABHD6—plasma membrane-bound enzyme). G3P can be directly converted to glycerol via the glycerol-3-phosphate phosphatase (G3PP) as a mechanism of gluco-detoxification. The triggering pathway of insulin secretion is shown in blue. Activation of FFAR1 further enhances insulin secretion, as shown in green.
2. The GL/NEFA Cycle in a Physiological Context
The GL/NEFA cycle is composed of lipogenic and lipolytic arms (Figure 1). Lipogenesis involves the consecutive formation of lysophosphatidic acid (LPA), phosphatidic acid (PA), DAG, and finally TAG from glycolysis-derived glycerol-3-phosphate (G3P) and Lc-CoA. The lipolysis arm of the cycle breaks down TAG to DAG and MAG, ultimately releasing glycerol [3]. As β-cells barely express glycerol kinase, glycerol leaves the cell through aquaglyceroporins [4][5]. Enzymes catalyzing lipogenesis and lipolysis reactions are not fully described but consist principally of redundant and compartmentalized acyltransferases (GPAT, AGPAT, DGAT) and lipases (ATGL, HSL, MAGL/ABHD6) [3][6]. As shown in INS-1E cells and human islets, glucotoxic conditions upregulate genes encoding enzymes of the GL/NEFA cycle [7], underlying the interplay between high glucose and intracellular lipid turnover.
Glucose stimulation induces lipogenesis by redirecting NEFA to esterification and by favoring de novo lipid synthesis [8]. In the β-cell, the latter pathway is marginal due to low expression of fatty acid synthase [9], and paradoxically, glucose promotes lipolysis [10][11][12][13]. This suggests that GL/NEFA cycling is implicated in β-cell activity. Mulder et al. first substantiated the role of the GL/NEFA cycle in β-cell function by showing that inhibition of lipolysis by the pan lipase inhibitor Orlistat impairs GSIS [14]. A few years later, Fex et al. showed that mice lacking HSL in the β-cells exhibit altered GSIS [15] and Attané et al. demonstrated that β-cell-specific ATGL knockout mice have lower insulinaemia and GSIS [16]. Collectively, these results underscore the role of the GL/NEFA cycle in general and its lipolytic arm in particular for proper β-cell function.
Intermediates of lipid metabolism, mainly from the GL/FFA cycle, have been put forward as coupling factors in GSIS. However, their exact nature and the mechanisms by which they modulate GSIS remain unclear [3]. Among those metabolites, DAG and MAG have attracted much of the attention. DAG is formed either during lipogenesis from PA or upon lipolysis from direct hydrolysis of TAG. According to the specificity of the biochemical reactions, different species of DAG may be produced. Lipin-mediated PA phosphohydrolase generates 1,2-DAG, while the hydrolysis of TAG by ATGL produces either 1,3-DAG or 2,3-DAG [17][18]. Among these DAG species, only 1,2-DAG qualifies as a signaling molecule by activating PKC, which further phosphorylates target proteins of the exocytotic machinery and induces actin remodeling [19]. Regarding MAG, it was shown to promote insulin granule exocytosis by binding to the exocytotic protein Munc13-1 [20][21]. The role of MAG in β-cells has been substantiated in ATGL knockout mice where exogenous MAG supply restores GSIS [16].
3. GL/NEFA Cycling in Type-2 Diabetes
Obesity may lead to β-cell dysfunction and potentially diabetes through the mechanisms of glucolipotoxicity described above. Potential β-cell failure is preceded by a compensatory phase during which β-cell mass and insulin secretory capacity are increased in order to compensate for obesity-induced insulin resistance [22][23]. Specifically, it was suggested that, in this context, GL/NEFA cycling is enhanced as an adaptive response [3]. When islets isolated from obese Zucker-Fatty rats are acutely exposed to stimulatory glucose and palmitate, GSIS is increased, along with NEFA esterification and lipolysis, while lipase inhibition reduces insulin secretion [24]. In the compensatory phase, enhanced GL/NEFA cycling would promote higher β-cell secretory capacity by increasing the production of coupling factors such as DAG and MAG. However, it has also been proposed by Prentki and colleagues that this cycle could serve as a path for excess fuel detoxification [3]. Indeed, converted to glycerol part of surplus glucose is no longer metabolized and leaves the β-cell through aquaglyceroporins, relieving the cell from some of the glucotoxic effects. The identification of a glycerol-3-phosphate phosphatase (G3PP) that converts G3P to glycerol without engaging the whole GL/NEFA cycle is supportive of this glycerol-mediated gluco-detoxification [25]. Similarly, esterification of NEFA from Lc-CoA and glycerol-3-phosphate into TAG would promote lipo-detoxification by preventing the formation of harmful lipid derived species.
The exact role of the GL/NEFA cycle in β-cell glucolipotoxicity remains unclear, and its contribution to the development of type-2 diabetes lacks in vivo evidence. In rodent models, it was observed that lipolysis and NEFA esterification were reduced in high-fat diet-induced obese mice exhibiting early and pre-diabetes, suggesting alteration of the GL/NEFA cycle already in the pre-diabetic stage [26]. Several other studies reported unbalanced GL/NEFA cycle in rodent models of type-2 diabetes, notably increased lipogenesis accompanied by reduced or unchanged lipolysis [3]. On the basis of studies in human islets and INS-1E cells, we recently reported that the GL/NEFA cycle plays a central role in the preservation of GSIS upon glucolipotoxic culture conditions [7]. Such a protective effect requires acute intracellular TAG mobilization, thanks to the adaptive response of GL/NEFA cycle gene expression [7]
This entry is adapted from the peer-reviewed paper 10.3390/ijms23010324
References
- Corkey, B.E.; Glennon, M.C.; Chen, K.S.; Deeney, J.T.; Matschinsky, F.M.; Prentki, M. A role for malonyl-CoA in glucose-stimulated insulin secretion from clonal pancreatic beta-cells. J. Biol. Chem. 1989, 264, 21608–21612.
- Brun, T.; Roche, E.; Kim, K.H.; Prentki, M. Glucose regulates acetyl-CoA carboxylase gene expression in a pancreatic beta-cell line (INS-1). J. Biol. Chem. 1993, 268, 18905–18911.
- Prentki, M.; Madiraju, S.R. Glycerolipid/free fatty acid cycle and islet beta-cell function in health, obesity and diabetes. Mol. Cell. Endocrinol. 2012, 353, 88–100.
- Noel, R.J.; Antinozzi, P.A.; McGarry, J.D.; Newgard, C.B. Engineering of glycerol-stimulated insulin secretion in islet beta cells. Differential metabolic fates of glucose and glycerol provide insight into mechanisms of stimulus-secretion coupling. J. Biol. Chem. 1997, 272, 18621–18627.
- Matsumura, K.; Chang, B.H.; Fujimiya, M.; Chen, W.; Kulkarni, R.N.; Eguchi, Y.; Kimura, H.; Kojima, H.; Chan, L. Aquaporin 7 is a beta-cell protein and regulator of intraislet glycerol content and glycerol kinase activity, beta-cell mass, and insulin production and secretion. Mol. Cell. Biol. 2007, 27, 6026–6037.
- Mulder, H.; Holst, L.S.; Svensson, H.; Degerman, E.; Sundler, F.; Ahren, B.; Rorsman, P.; Holm, C. Hormone-sensitive lipase, the rate-limiting enzyme in triglyceride hydrolysis, is expressed and active in beta-cells. Diabetes 1999, 48, 228–232.
- Oberhauser, L.; Jiménez-Sánchez, C.; Madsen, J.G.S.; Duhamel, D.; Mandrup, S.; Brun, T.; Maechler, P. Glucolipotoxicity promotes the capacity of the glycerolipid/free fatty acid cycle supporting the secretory response of pancreatic beta-cells. Diabetologia 2022, in press.
- Sandberg, M.B.; Fridriksson, J.; Madsen, L.; Rishi, V.; Vinson, C.; Holmsen, H.; Berge, R.K.; Mandrup, S. Glucose-induced lipogenesis in pancreatic beta-cells is dependent on SREBP-1. Mol. Cell. Endocrinol. 2005, 240, 94–106.
- Brun, T.; Roche, E.; Assimacopoulos-Jeannet, F.; Corkey, B.E.; Kim, K.H.; Prentki, M. Evidence for an anaplerotic/malonyl-CoA pathway in pancreatic beta-cell nutrient signaling. Diabetes 1996, 45, 190–198.
- Nolan, C.J.; Madiraju, M.S.R.; Delghingaro-Augusto, V.; Peyot, M.L.; Prentki, M. Fatty Acid Signaling in the -Cell and Insulin Secretion. Diabetes 2006, 55 (Suppl. S2), S16–S23.
- Fex, M.; Mulder, H. Lipases in the pancreatic beta-cell: Implications for insulin secretion. Biochem. Soc. Trans. 2008, 36 Pt 5, 885–890.
- Chlouverakis, C. The action of glucose on lipolysis. Metabolism 1967, 16, 469–472.
- Winzell, M.S.; Strom, K.; Holm, C.; Ahren, B. Glucose-stimulated insulin secretion correlates with beta-cell lipolysis. Nutr. Metab. Cardiovasc. Dis. 2006, 16 (Suppl. S1), S11–S16.
- Mulder, H.; Yang, S.; Winzell, M.S.; Holm, C.; Ahren, B. Inhibition of lipase activity and lipolysis in rat islets reduces insulin secretion. Diabetes 2004, 53, 122–128.
- Fex, M.; Haemmerle, G.; Wierup, N.; Dekker-Nitert, M.; Rehn, M.; Ristow, M.; Zechner, R.; Sundler, F.; Holm, C.; Eliasson, L.; et al. A beta cell-specific knockout of hormone-sensitive lipase in mice results in hyperglycaemia and disruption of exocytosis. Diabetologia 2009, 52, 271–280.
- Attane, C.; Peyot, M.L.; Lussier, R.; Poursharifi, P.; Zhao, S.; Zhang, D.; Morin, J.; Pineda, M.; Wang, S.; Dumortier, O.; et al. A beta cell ATGL-lipolysis/adipose tissue axis controls energy homeostasis and body weight via insulin secretion in mice. Diabetologia 2016, 59, 2654–2663.
- Eichmann, T.O.; Lass, A. DAG tales: The multiple faces of diacylglycerol—Stereochemistry, metabolism, and signaling. Cell. Mol. Life Sci. 2015, 72, 3931–3952.
- Eichmann, T.O.; Kumari, M.; Haas, J.T.; Farese, R.V., Jr.; Zimmermann, R.; Lass, A.; Zechner, R. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J. Biol. Chem. 2012, 287, 41446–41457.
- Trexler, A.J.; Taraska, J.W. Regulation of insulin exocytosis by calcium-dependent protein kinase C in beta cells. Cell Calcium 2017, 67.
- Zhao, S.; Mugabo, Y.; Iglesias, J.; Xie, L.; Delghingaro-Augusto, V.; Lussier, R.; Peyot, M.L.; Joly, E.; Taib, B.; Davis, M.A.; et al. alpha/beta-Hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell Metab. 2014, 19, 993–1007.
- Sheu, L.; Pasyk, E.A.; Ji, J.; Huang, X.; Gao, X.; Varoqueaux, F.; Brose, N.; Gaisano, H.Y. Regulation of insulin exocytosis by Munc13-1. J. Biol. Chem. 2003, 278, 27556–27563.
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontes, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta 2010, 1801, 289–298.
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 2006, 116, 1802–1812.
- Nolan, C.J.; Leahy, J.L.; Delghingaro-Augusto, V.; Moibi, J.; Soni, K.; Peyot, M.L.; Fortier, M.; Guay, C.; Lamontagne, J.; Barbeau, A.; et al. Beta cell compensation for insulin resistance in Zucker fatty rats: Increased lipolysis and fatty acid signalling. Diabetologia 2006, 49, 2120–2130.
- Mugabo, Y.; Zhao, S.; Seifried, A.; Gezzar, S.; Al-Mass, A.; Zhang, D.; Lamontagne, J.; Attane, C.; Poursharifi, P.; Iglesias, J.; et al. Identification of a mammalian glycerol-3-phosphate phosphatase: Role in metabolism and signaling in pancreatic beta-cells and hepatocytes. Proc. Natl. Acad. Sci. USA 2016, 113, E430–E439.
- Peyot, M.L.; Pepin, E.; Lamontagne, J.; Latour, M.G.; Zarrouki, B.; Lussier, R.; Pineda, M.; Jetton, T.L.; Madiraju, S.R.; Joly, E.; et al. Beta-cell failure in diet-induced obese mice stratified according to body weight gain: Secretory dysfunction and altered islet lipid metabolism without steatosis or reduced beta-cell mass. Diabetes 2010, 59, 2178–2187.
This entry is offline, you can click here to edit this entry!