Platelet-rich thrombi formed in vivo in mice have been shown to be composed of a core and shell region. Different pathways regulate the formation and stability of these two regions and understanding them may result in new ways to treat arterial thrombosis. The distinguishing feature between these two regions is the absence of fibrin in the shell, which indicates that in-vitro flow-based assays over thrombogenic surfaces, in the absence of coagulation, can be used to resemble this region. Glycoprotein VI (GPVI) is a platelet immunoglobulin receptor which is known as a receptor for collagen and has been shown to contribute to the stability of platelet aggregates on collagen at high shear. In recent years GPVI has also been shown to be a receptor for fibrin and fibrinogen. Since the activation of GPVI by fibrinogen is also dependent on integrin αIIbβ3, with the interplay of the two receptors driving platelet adhesion and activation, blocking signalling pathways common to both receptors may have a greater antithrombotic effect than blocking GPVI alone. Therefore in this study, we have investigated the contribution of Syk tyrosine kinase, which plays a critical role in signalling by integrin αIIbβ3 and GPVI, in the stability of platelet aggregates formed on collagen or atherosclerotic plaque homogenate at arterial shear (1000 s-1). The results show that Syk regulates thrombus stability in the absence of fibrin. The fact that inhibitors of Syk are currently used for the treatment of patients with refractory immune thrombocytopenia (ITP) without increasing the risk of bleeding, indicate that Syk inhibitors may represent a new class of antiplatelet agent with reduced bleeding risk compared to current drugs.
1. Introduction
Platelet-rich thrombi formed in vivo in mice have been shown to be composed of a core and shell region in both the arterial and venous microcirculation [
1]. In the core region, platelets and fibrin are densely packed, and permeability is heavily restricted [
2], necessitating the use of a fibrinolytic to dissolve the platelet aggregate [
3,
4]. The outer, more permeable, shell region however, consists of weakly activated and loosely packed platelets and does not contain fibrin [
2], suggesting that a different strategy is needed to promote disruption of this region and thus help to prevent the build-up of an occlusive thrombus.
Glycoprotein VI (GPVI) is a platelet immunoglobulin receptor which is known as a receptor for collagen [
5], but in recent years has also been shown to be a receptor for fibrin and fibrinogen, among other predominantly charged ligands [
6]. In vitro flow studies have shown that GPVI contributes to the stability of newly formed platelet aggregates on collagen at high shear through use of the blocking anti-GPVI Fab ACT017, which has more recently been named glenzocimab [
4]. Since the activation of GPVI by fibrinogen is also dependent on integrin αIIbβ3, with the interplay of the two receptors driving platelet adhesion and activation [
7]; this suggests that blocking signalling pathways common to both receptors may have a greater antithrombotic effect than blocking GPVI alone.
The tyrosine kinase Syk plays a critical role in signalling by integrin αIIbβ3 and GPVI [
8,
9] and an inhibitor of Syk, fostamatinib, is clinically used for the treatment of patients with refractory immune thrombocytopenia (ITP) without increasing the risk of bleeding despite the marked reduction in platelet count in this patient group [
10]. Moreover, a retrospective analysis provides evidence of a notably low incidence of thrombotic events in patients treated with the Syk inhibitor [
10]. Thus, Syk inhibitors represent a new class of antiplatelet agent with reduced bleeding risk compared to current drugs. The involvement of Syk in thrombus formation and thrombus growth and stabilisation suggests that inhibitors will be effective against multiple stages in thrombus formation.
2. Thrombin Stimulates Sustained Phosphorylation of Syk via Integrin αIIbβ3 during Platelet Aggregation
The outer shell of an arterial thrombus is composed of aggregated platelets held together by the interaction of fibrinogen with integrin αIIbβ3, with activation of the integrin mediated by the autocrine feedback mediators, ADP and TxA
2 [
18,
19]. It has been proposed that the two mediators diffuse from the highly activated platelets in the thrombus core region to support platelet aggregation in the shell [
20,
21]. In this model, it is unclear how the activation of the integrin is maintained over time as the levels of the two mediators decline due to exhaustion of intracellular stores and reduced activation of phospholipase A
2. One potential explanation is that integrin activation is maintained by a positive feedback pathway mediated through the activation of αIIbβ3 and GPVI by fibrinogen [
7]. If this is the case, the activation of Syk during platelet aggregation should be sustained.
To investigate this, we activated platelets in suspension by thrombin which signals through the G
q protein-coupled receptors, PAR-1 and PAR-4 [
22]. Thrombin was selected for the experiments because it is the most powerful G
q protein-coupled receptor ligand in platelets. We performed the experiments in the absence or presence of the αIIbβ3 antagonist, eptifibatide, to establish the role of the integrin in mediating Syk activation.
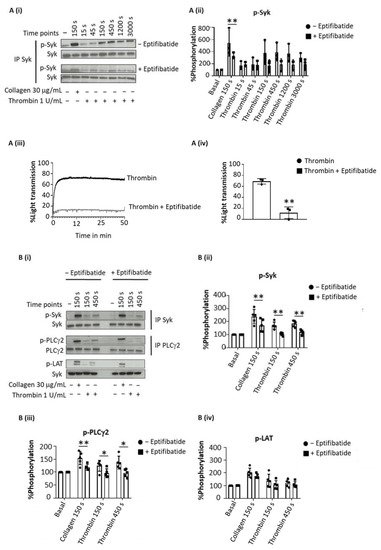
Thrombin evoked sustained tyrosine phosphorylation of Syk over 150–3000 s (as concluded from 4G10 staining after Western blotting), which was inhibited by eptifibatide at all time points (Figure 1(Ai–iv)). In contrast, collagen-induced tyrosine phosphorylation of Syk, was reduced but not blocked by eptifibatide (Figure 1(Ai,ii)). Eptifibatide also blocked the increase in tyrosine phosphorylation of LAT and PLCγ2 by thrombin but not by collagen (Figure 1(Bi–iv)).
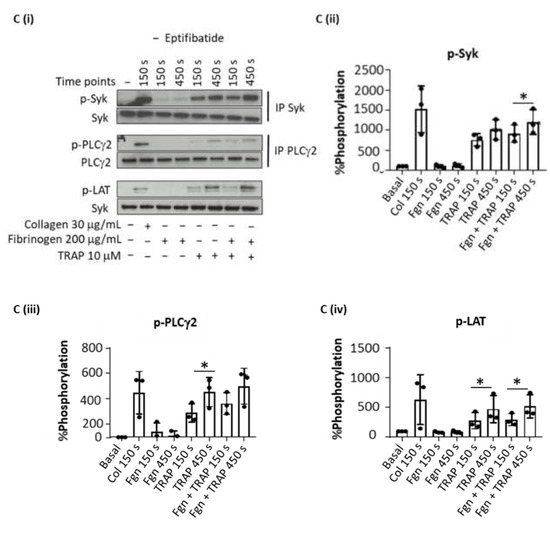
Figure 1. Sustained Syk phosphorylation in thrombin-stimulated platelet is integrin-dependent. Washed platelets at 5 × 108/mL were stimulated with Horm collagen (30 µg/mL), thrombin (1 U/mL), TRAP (10 µM) or fibrinogen (200 µg/mL) in the presence or absence of eptifibatide (9 µM). Activation was stopped at stated time after addition of the agonist. (A) Representative blot of Syk tyrosine phosphorylation from 6 donors. (Ai) Whole cell lysates were immunoprecipitated with anti-Syk mAb and probed with anti-phospho-tyrosine mAb 4G10; (Aii) Percentage of Syk tyrosine phosphorylation, based on Western blots, from 6 donors (mean ± s.d.); (Aiii) Aggregation trace of platelets stimulated with thrombin (1 U/mL), representative of 3 donors, and (Aiv) graph showing aggregation as % light transmission (mean ± s.d.) after 50 min of agonist stimulation. (B) Representative Western blot (Bi), and calculated percentage of Syk (Bii), PLCγ2 (Biii) and LAT (Biv) phosphorylation in response to thrombin (Thromb) from 5 donors (mean ± s.d.). Horm collagen (Col) was used as positive control. (C) Representative blot (Ci); mean ± s.d. of Syk (Cii), PLCγ2 (Ciii) and LAT (Civ) phosphorylation in response to collagen (Col), TRAP or fibrinogen (Fgn) from 3 donors. * p < 0.05, ** p < 0.005, one-tailed paired Student’s t-test. Where stated, proteins were immunoprecipitated (IP).
To rule out a possible role of fibrin generation by thrombin in these experiments, the studies were repeated with the PAR-1 peptide agonist TRAP (Figure 1(Ci)). TRAP also induced a sustained phosphorylation of Syk, LAT and PLCγ2 (Figure 1(Cii–iv)), whereas fibrinogen had no effect, as expected.
These data show a sustained activation of Syk downstream of integrin αIIbβ3 raising the possibility that this contributes to the maintenance of integrin activation during platelet aggregation.
3. Inhibition of Syk on Collagen and Plaque Homogenate Causes Thrombus Shell Instability
Experiments were designed to investigate the contribution of Syk to GPVI-dependent thrombus stability. In these experiments, recalcified blood was perfused for 7 min at arterial shear (1000 s
−1) over Horm collagen or human plaque material. Both ligands stimulate platelet activation through GPVI [
5,
12]. The thrombin inhibitor, PPACK, was used to prevent fibrin formation.
Aggregates were post-perfused for 10 min with vehicle (DMSO) or the Syk inhibitor, PRT-060318, at a concentration (10 µM) that has been shown to be effective in whole blood [
23,
24].
Heparin was added to the post-perfusion buffer to prevent residual formation of fibrin. Thrombus disaggregation was monitored by taking brightfield images at 60 s intervals.
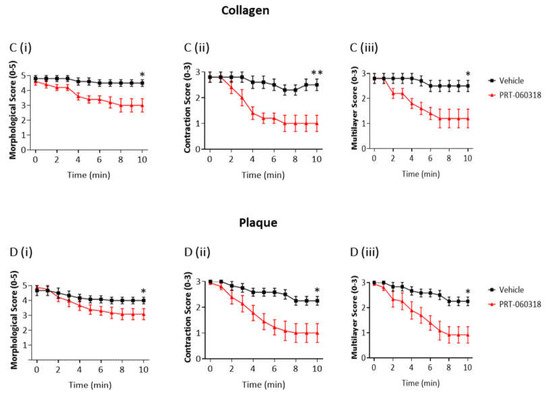
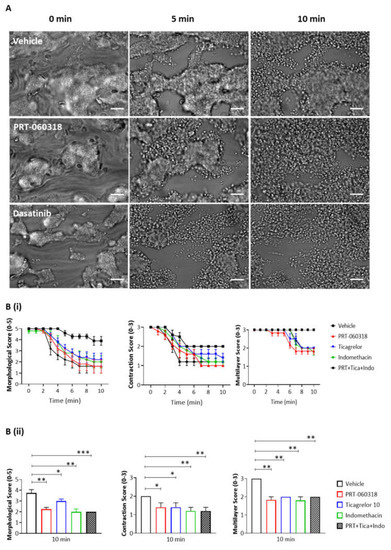
Aggregates formed after 7 min of blood perfusion on both collagen and plaque material were tightly contracted and consisted of multiple platelet layers as illustrated in Figure 2A,B. Post-perfusion with vehicle for 10 min had a minor effect on the architecture, as reflected by the small reduction in morphological and contraction scores (Figure 2(Ci,ii,Di,ii)). The aggregates were still composed of multiple layers of platelets as shown by the multilayer score (Figure 2(Ciii,Diii)). In contrast, perfusion with PRT-060318 on both surfaces promoted aggregate breakdown which could be clearly seen by eye (Figure 2A,B) and was confirmed by measurement of morphology (Figure 2(Ci,Di)). The increased breakdown could be detected within 3 min as shown by the reduction in the contraction score at this time (Figure 2(Cii,Dii)). The contraction score had return to base-line by 6–10 min. The loss of stability led to a slow detachment of platelets eventually forming a monolayer on the surface (Figure 2(Ciii,Diii)).
Figure 2. Inhibition of Syk causes loss of contraction and platelet adhesion on collagen and plaque material. Recalcified blood from 6 donors was perfused for 7 min on Horm collagen and plaque at room temperature at a shear rate of 1000 s
−1. (
A,
B) Representative images of aggregates formed on collagen (
A) and plaque (
B), taken during post-perfusion at times 0 and 10 min with vehicle or with PRT-060318 (
n = 6). Scale bar = 100 µm. (
C,
D) Disaggregation on collagen (
C) or on plaque (
D) was monitored by taking brightfield images every 60 sec and measured using morphological (
Ci,
Di), contraction (
Cii,
Dii) and multilayer scores (
Ciii,
Diii) [
16]. Aggregates were post-perfused with rinse buffer ± vehicle (DMSO) or the Syk inhibitor PRT-060318 (10 µM). Data are shown as mean ± s.e.m. *
p < 0.05, **
p < 0.005.
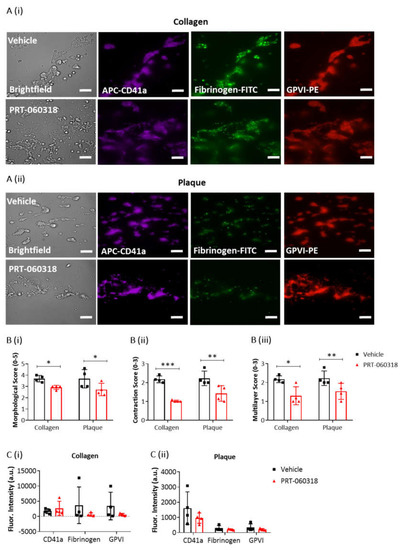
To further explore the effect of Syk inhibition on thrombus characteristics, recalcified blood was triple labelled with PE-anti-GPVI mAb, APC-anti-CD41a mAb and FITC-anti-fibrinogen prior to perfusion over collagen and plaque, and then post-perfused for 3 min in the presence of buffer or PRT-060318 (Figure 3(Ai,ii)). PPACK and heparin were included as above to prevent thrombin formation. At this early time, perfusion with the Syk inhibitor caused a significant impairment in morphological, contraction and multilayer scores (Figure 3B). Fluorescence images of the aggregates indicated a reduced labelling of fibrinogen and GPVI staining in the presence of PRT-060318 although this did not reach significance (Figure 3C). Imaging was not performed at later timepoints due to the reduction in intensity through loss of platelets.
Figure 3. Platelet activation imaging of thrombi postperfused under condition of Syk inhibition. Recalcified blood samples labelled with APC-αCD41a mAb (purple), FITC-fibrinogen (green) and PE-αGPVI mAb (red) was perfused for 7 min over immobilised collagen or plaque material at room temperature and arterial shear rate (1000 s
−1). Post-perfusion for 3 min was with rinse buffer containing vehicle (DMSO) or Syk inhibitor PRT-060318 (10 µM). (
A) Representative microscopic images, showing remaining aggregates on collagen (
Ai) or plaque (
Aii) after 3 min of post-perfusion with vehicle or PRT-060318 (
n = 4 donors). (
B) Graphs showing scores of thrombus morphology (
Bi), thrombus contraction (
Bii) and thrombus multilayer (
Biii) [
16]. (
C) Graphs of fluorescence intensity (arbitrary units, a.u.) of platelet aggregates on collagen (
Ci) or plaque material (
Cii) fixed after 3 min of post-perfusion. Scale bar = 50 µm. Data are shown as mean ± s.d., *
p < 0.05, **
p < 0.005, ***
p < 0.0005, one-tailed Student’s paired
t-test.
Taken together, the above results show that post-inhibition of Syk promotes disaggregation of the preformed thrombi on surfaces of collagen or plaque which eventually leads to the formation of a platelet monolayer.
4. Inhibition of Syk Promotes Thrombus Breakdown at 37 °C
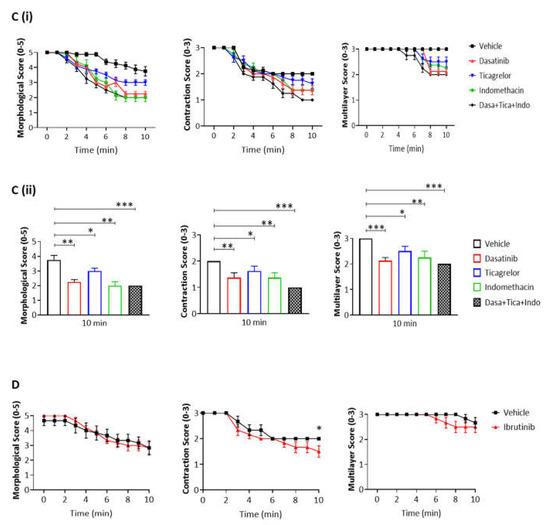
We next asked if the contribution of Syk to thrombus shell architecture is also seen at the body temperature of 37 °C. The thrombi formed at 37 °C were larger and more tightly packed than those formed at room temperature (Figure 4A). In the presence of PRT-060318, the aggregates could be seen to loosen and to spread out from approximately 2 min of perfusion with the Syk inhibitor relative to vehicle (Figure 4(Bi,ii)). However, the majority of platelets was retained in the aggregate for up to 5 min and only at later times could they be seen to detach resulting in a significant decrease in the multilayer score (Figure 4(Bi,ii)).
Figure 4. Inhibition of Syk causes the same extent of disaggregation observed with antagonists of adenosine diphosphate (ADP) and thromboxane A2 (TxA2). Recalcified blood was perfused for 7 min on Horm collagen at 37 °C at arterial shear (1000 s−1), and post-perfused for 10 min with rinse buffer with vehicle (DMSO) or inhibitor of Syk, Src or Btk (PRT-060318 10 µM, dasatinib 10 µM or ibrutinib 7 µM, respectively). (A) Representative images of aggregates at 0, 5, and 10 min of post-perfusion with vehicle, PRT-060318 (n = 5) or dasatinib (n = 6). (B–D) Graphs showing extent of disaggregation, as measured from scores of thrombus morphology, contraction and multilayer,16 at every 60 s (Bi,Ci,D) or at 10 min (Bii,Cii) of post-perfusion with either vehicle, PRT-060318 (PRT) (Bi,ii), dasatinib (Ci,ii) or ibrutinib (D), alone or combined with indomethacin (Indo, 10 µM) and ticagrelor (Tica, 10 µM). Scale bar = 50 µm. Data are shown as mean ± s.e.m. * p < 0.05, ** p < 0.005, *** p < 0.0005, one-tailed paired Student’s t-test.
This shows that Syk contributes to thrombus stability at both room temperature and at 37 °C temperature despite the increased stability of the aggregates at the higher temperature.
5. Inhibition of Src and Secondary Agonists but Not Btk Promotes Thrombus Breakdown
We then extended the studies at 37 °C to investigate the role of Src and Btk tyrosine kinases in aggregate stability, alongside the secondary mediators ADP and TxA
2. Src kinases lie both upstream and downstream of Syk in the GPVI signalling cascade, and Btk lies downstream of both kinases [
25].
As observed with the Syk inhibitor PRT-060318, the Src inhibitor dasatinib (10 µM) induced an increase in aggregate breakdown at 2 min of post-perfusion (
Figure 4(Ci,ii)), with a marked loss of aggregate architecture after 6 min of post-perfusion similar to that observed in the presence of PRT-060318 (
Figure 4B,C). In contrast, the Btk inhibitor ibrutinib (7 µM), at a concentration that is effective in plasma [
26] caused only a mild decrease in the contractile score and no apparent change in morphology (
Figure 4D). The much weaker effect may be due to redundancy with the Tec family kinase, Tec, or the later role of Btk in the GPVI signalling cascade.
The dependency of thrombus stability on the secondary mediators, ADP and TxA2, was investigated using the P2Y12 receptor antagonist ticagrelor (10 µM) and cyclooxygenase inhibitor indomethacin (10 µM), respectively. The two inhibitors had a similar effect on thrombus stability to that of PRT-060318 and dasatinib (Figure 4(Bii,Cii)). Moreover, the combination of indomethacin, ticagrelor and PRT-060318 caused only a slight increase in disaggregation suggesting that the two mediators work in concert with Syk to promote aggregation (Figure 4(Bii,Cii)). This could reflect either synergy of intracellular signals or a role of Syk in stimulating release of ADP and TxA2.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23010493