Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Multiple Myeloma (MM) is a genetically complex and heterogeneous hematological cancer that remains incurable despite the introduction of novel therapies in the clinic. The surprising finding that MM cells present rampant genomic instability has ignited concerted efforts to understand its origin and exploit it for therapeutic purposes.
- Multiple Myeloma
- genomic instability
- replicative stress
1. Introduction
Multiple Myeloma (MM) is a hematological malignancy characterized by the accumulation of malignant plasma cells in the bone marrow and, during the progression of the disease, in the peripheral blood and other extramedullary sites. MM is associated with the production of immunoglobulin monoclonal proteins (M-protein), which are used as a reliable biomarker for the number of tumor cells in the body and are measured for clinical purposes in the serum and/or urine [1]. The accumulation of cancer cells is responsible for some of the clinical manifestations of the disease, including anemia, bone marrow failure, and bone destruction, which often results in hypercalcemia, that aggravates renal insufficiency, almost always occurring with the increase in the serum monoclonal (M) protein [2]. MM presents a median age at diagnosis of 69 years. In the USA, MM is the second most frequent hematological cancer after lymphomas, accounting for 18% of hematological malignancies, 1.8% of all new cancer cases, and 2% of all cancer deaths (Stat Fact Sheets SEER. Myeloma. http://seer.cancer.gov/statfacts/html/mulmy.html. accessed on 12 November 2021; [3]). MM is always preceded by a premalignant state, monoclonal gammopathy of undetermined significance (MGUS), which, in a small subset of patients, evolves towards an indolent form, smoldering myeloma (SMM), and finally to MM [4].
Molecular profiling technologies, including fluorescence in situ hybridization (FISH), microarray gene expression profiling (GEP), array comparative genomic hybridization (CGH), single-nucleotide polymorphism (SNP) arrays, and, recently, next generation sequencing (NGS), have brought a fairly comprehensive view on the genomic landscape of MM [5]. This knowledge has led to the identification of several molecular drivers triggering MM initiation and progression but has, disappointingly, failed to provide effective therapeutic targets or even biomarkers that could be used to dictate treatment strategies [5]. Conversely, other approaches have proven quite effective in delaying the progression of the disease, including treatments addressing MM non-oncogenic addictions [6], the interactions of MM cells with the microenvironment, and the presence of surface markers on MM plasma cells. Accordingly, the introduction of proteasome inhibitors, which hamper protein clearance inside plasma cells already overwhelmed by the production of immunoglobulins [7][8]; of lenalidomide-based treatments, which target for degradation essential factors for MM cell survival, including the Ikaros family zinc finger proteins 1 and 3 (IKZF1 and IKZF3), but also interfere with angiogenesis [9][10]; and most recently, of anti-CD-38 monoclonal antibodies [11][12][13][14], which have led to an important improvement in the survival of MM patients. However, despite this impressive evolution of treatment regimens, MM remains an incurable disease [15], warranting new therapeutic strategies. Notably, patients presenting with the unfavorable disease have benefited the least from the introduction of new drugs, thus representing still a highly vulnerable population [15].
Genomic instability has been recognized as a driving force in epithelial cancers [16], which, on one side, is an asset for cancer cells, broadening their evolutionary pool, but on the other is a liability. Conversely, blood cancers present a much more stable genome when compared with epithelial cancers [17], suggesting that genomic instability, and the ability to exploit it, may not represent a feature of hematological tumors. Yet, recent evidence accrued, especially in MM, would suggest otherwise.
2. Genomic Instability in Cancer
The existence of genomic instability in cancer has been debated for decades. It was unclear whether the multitude of genomic lesions present in cancer genomes resulted from a sudden, short-lived “big bang” [18][19][20][21], disrupting genome integrity, or was instead the result of a steadier, incremental accumulation of genetic lesions, reflecting an inherent, and cancer-specific, enhanced tendency to acquire genomic alterations over time [16]. It is increasingly apparent that both mechanisms could occur in cancer cells, with the latter labeled as genomic instability [16], which is now included among the enabling characteristics of cancer [22].
Why did it take so long to solve this debate? One possible explanation lies with the assay, as even measuring genomic instability is challenging. A common misconception equates genomic instability with the number of genetic lesions present at a defined point and time in a cell, hence, a state and not a rate (as aptly noted by Bert Vogelstein and collaborators several years ago [23]). As such, the assays currently used to define genomic alterations are descriptive of the cell (and the genome) status at a certain time and do not register ongoing changes. One potential assay, which may more precisely register instability, albeit not perfectly, is the measure of phosphorylated H2AX (γH2AX), which may quantify at least a specific subtype of instability. When γH2AX foci are more intense and numerous in cancer cells when compared with the corresponding normal tissues [24][25][26], this phenomenon may record an ongoing disruption of DNA. It should be noted, however, that this assay measures just a subset of all the potential phenotypes associated with genomic instability. For example, it does not measure the increased mutational load emerging from mismatch repair or chromosomal instability elicited by mitotic disruption.
As for the causes of genomic instability, in hereditary tumors, germline mutations in the so-called caretaker genes, such as BRCA1 and BRCA2, have been reported and linked to genomic instability [22].
However, most cancers are sporadic. In this setting, what is the cause of genomic instability? Based on observations gathered at the transition between benign and malignant cancers [27][28][29], a hypothesis has been put forward which blames the very genes engaged early in cancer development as the major culprits of genomic instability in sporadic cancers. The theory goes that oncogenes activation (or inactivation of tumor suppressors which control the cell cycle progression) deregulates DNA replication [29], eliciting replicative stress, which, in turn, causes genomic instability.
3. Replicative Stress as a Key Driver of Genomic Instability in Cancer
DNA replication is a tightly regulated process, as even a modest disruption may lead to fatal consequences. As such, cells devote a considerable amount of energy and an exquisitely fine-tuned molecular machine and several checkpoints to ensure its accuracy [30].
What, then, is replicative stress? The term broadly defines impediments to DNA replication, which result in the stalling and eventual collapse of the replication forks, causing DNA damage [31]. The causes of replication fork stalling are several, ranging from inappropriate origin firing to the presence of unresolved DNA secondary structures, even exhaustion of the nucleotide pools available for DNA synthesis or the presence of DNA–RNA hybrid intermediates [32] (Figure 1). An intriguing mechanism leading to fork stalling relates to the presence of highly expressed genes, which challenge replisome progression, triggering replication–transcription machinery collisions [32][33][34]. More specifically, DNA replication and transcription machinery during the S phase share the same DNA template, and defects in their coordination generate transcription–replication conflicts (TRCs). These conflicts occur when the replication forks encounter the RNA polymerase, thus stalling. This favors the transient formation of R-loops, nucleic acid structures composed of a DNA–RNA hybrid and the displaced non-template single-stranded DNA (ssDNA) [35][36], which is particularly prone to breaks due to the high accessibility of the ssDNA to metabolites, reactive oxygen species (ROS), and nucleases.
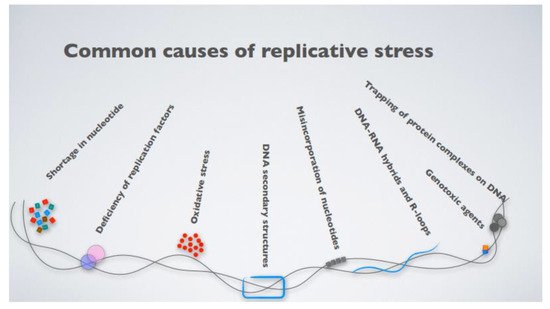
Figure 1. The most common causes contributing to cellular replicative stress.
The induction of DNA double-strand breaks (DSBs) following activation of many oncogenes is the result of their ability to lead to the pervasive formation of stalled forks which eventually collapse [31]. Several mechanisms can contribute to oncogene-induced DNA replicative stress. For example, activation of oncogenic RAS driving proliferation and cell growth increases the number of active replicons and leads to asymmetric fork progression [28]. It should be noted that RAS, like other oncogenes, might elicit the fork stalling through additional mechanisms. RAS, for example, also suppresses nucleotide metabolism by downregulating the ribonucleotide reductase subunit M2 (RRM2), a rate-limiting protein in dNTP synthesis, and causing dNTP pool depletion and premature termination of replication forks [37]. Moreover, RAS overexpression leads to increased global transcription activity to face the additional demands stemming from the increased proliferation [38], which is then linked to replicative stress due to R-loop accumulation and increased expression of the general transcription factor TBP [38].
The oncogene MYC, as well, induces replicative stress through various mechanisms. MYC overexpression causes the direct deregulation of DNA replication dynamics due to interaction with members of the DNA replication machinery [39][40][41][42]. Additionally, MYC is a transcription factor that controls the expression of a large fraction of cellular genes linked to cell cycle control. MYC may promote cell cycle progression and replicative stress through indirect mechanisms, by regulating the expression of genes involved in cellular proliferation [43] and DNA replication, including the majority of genes involved in the nucleotide biosynthesis pathway [44]. Finally, MYC also induces replicative stress through the generation of TRCs [44].
There are also additional mechanisms by which oncogenes may induce replicative stress. Aberrant oncogene expression results in the loss of redox homeostasis with the unbridled generation of ROS [45]. Accumulation of ROS leads to the formation of 8-oxoguanine, and the resulting oxidative DNA damage causes the replication fork to stall at lesions, ultimately promoting replicative stress in cancer cells [42][46][47].
Finally, the induction of DNA DSBs can also be observed when tumor suppressors are lost. The deregulated activity of pRB, p53, p16, and p14ARF have been linked to replicative stress through various mechanisms that result in the promotion of G1-S transition [48].
4. Genomic Instability and Replicative Stress in Multiple Myeloma
So, how does this model also fit a slow-growing hematological cancer like multiple myeloma?
Very surprisingly, a few years ago, researchers and others discovered that MM cell lines, as well as patient cells, display exceedingly high levels of rampant DNA damage and DSBs, as assessed by the presence of γH2AX foci in the absence of exogenous stressors [49][50][51][52][53][54]. DNA damage is absent in normal plasma cells or B-cells but appears in MGUS samples and increases in MM patient samples and almost all reported MM cell lines [49][50][54]. This genomic instability was linked to replicative stress since MM cells with ongoing DNA damage demonstrated positivity to a panel of replicative stress markers, including the phosphorylated form of RPA32, a subunit of replication protein A (RPA) [50]. Accordingly, cell cycle regulatory genes and genes related to DNA replication were significantly altered in MM cells. Importantly, the cells of a subset of MM patients presented with a signature of chromosomal instability and increased expression of replicative genes; these patients presented a poor prognosis [50].
What could be the cause of this replicative stress? MYC has been long known as a crucial gene in MM progression [55][56][57]. Surprising recent observations [58][59][60][61], however, point to a role for MYC in the early phases of MM. Indeed, patients whose cancer cells presented with increased replicative stress showed enhanced expression of the oncogene MYC. The silencing of MYC in MM cells presenting DNA damage reduced the number of γH2AX foci, whereas MYC overexpression in the U266 MM cell line, endowed with low levels of DNA damage and no c-MYC genomic rearrangements nor MYC overexpression, triggered DSBs [50]. In all, these results suggest that MYC, a crucial gene in MM, fosters replicative stress in these cells.
It remains mysterious how a slow-growing, tumor-like MM could present such a high degree of replicative stress, despite the overexpression of the MYC oncogene. Likely, some of the mechanisms included in Figure 1 are not per se linked to enhanced proliferation and may elicit replicative stress, and thus genomic instability, in the absence of an apparent increased proliferation.
While a large array of oncogenes is genetically altered and/or overexpressed in MM, it is tempting to speculate that at least a few of them may be causative of replicative stress in this disease, including cyclins, present at early-occurring chromosomal translocations or overexpressed in the absence of clear genetic lesions, or the oncogenes MAF and MAFB [62]. Notwithstanding, the significant association between MYC expression and replicative stress, and the absence of replicative stress in a few MM cell lines without c-MYC overexpression, all point to a crucial role for this gene in triggering replicative stress in this disease.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14010025
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; van Duin, M.; Sonneveld, P.; Mateos, M.-V.; Gay, F.; Anderson, K.C. Multiple Myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046.
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group Updated Criteria for the Diagnosis of Multiple Myeloma. Lancet Oncol. 2014, 15, e538–e548.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30.
- Antonio, P.; Kenneth, A. Multiple Myeloma. N. Engl. J. Med. 2011, 364, 1046–1060.
- Pawlyn, C.; Davies, F.E. Toward Personalized Treatment in Multiple Myeloma Based on Molecular Characteristics. Blood 2019, 133, 660–675.
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of Cancer Therapy: Oncogene and Non-Oncogene Addiction. Cell 2009, 136, 823–837.
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The Proteasome Load versus Capacity Balance Determines Apoptotic Sensitivity of Multiple Myeloma Cells to Proteasome Inhibition. Blood 2009, 113, 3040–3049.
- Aronson, L.I.; Davies, F.E. DangER: Protein OvERload. Targeting Protein Degradation to Treat Myeloma. Haematologica 2012, 97, 1119–1130.
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305.
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G., Jr. The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science 2014, 343, 305–309.
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219.
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331.
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115.
- Minnie, S.A.; Hill, G.R. Immunotherapy of Multiple Myeloma. J. Clin. Investig. 2020, 130, 1565–1575.
- Binder, M.; Nandakumar, B.; Rajkumar, S.V.; Kapoor, P.; Buadi, F.K.; Dingli, D.; Lacy, M.Q.; Gertz, M.A.; Hayman, S.R.; Leung, N.; et al. Mortality Trends in Multiple Myeloma after the Introduction of Novel Therapies in the United States. Leukemia 2021.
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability—An Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228.
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218.
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.-C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated Copy Number Evolution and Clonal Stasis in Triple-Negative Breast Cancer. Nat. Genet. 2016, 48, 1119–1130.
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang Model of Human Colorectal Tumor Growth. Nat. Genet. 2015, 47, 209–216.
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40.
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic Instability in Colorectal Cancers. Nature 1997, 386, 623–627.
- Kuo, L.J.; Yang, L.-X. Gamma-H2AX—A Novel Biomarker for DNA Double-Strand Breaks. In Vivo 2008, 22, 305–309.
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. ΓH2AX and Cancer. Nat. Rev. Cancer 2008, 8, 957–967.
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA Damage Checkpoint and Genomic Instability in Human Precancerous Lesions. Nature 2005, 434, 907–913.
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-Induced Senescence Is Part of the Tumorigenesis Barrier Imposed by DNA Damage Checkpoints. Nature 2006, 444, 633–637.
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-Induced Senescence Is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature 2006, 444, 638–642.
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355.
- Nyberg, K.A.; Michelson, R.J.; Putnam, C.W.; Weinert, T.A. Toward Maintaining the Genome: DNA Damage and Replication Checkpoints. Annu. Rev. Genet. 2002, 36, 617–656.
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448.
- Zeman, M.K.; Cimprich, K.A. Causes and Consequences of Replication Stress. Nat. Cell Biol. 2014, 16, 2–9.
- Sulli, G.; Di Micco, R.; di Fagagna, F.A. Crosstalk between Chromatin State and DNA Damage Response in Cellular Senescence and Cancer. Nat. Rev. Cancer 2012, 12, 709–720.
- Bermejo, R.; Lai, M.S.; Foiani, M. Preventing Replication Stress to Maintain Genome Stability: Resolving Conflicts between Replication and Transcription. Mol. Cell 2012, 45, 710–718.
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618.
- Belotserkovskii, B.P.; Tornaletti, S.; D’Souza, A.D.; Hanawalt, P.C. R-Loop Generation during Transcription: Formation, Processing and Cellular Outcomes. DNA Repair 2018, 71, 69–81.
- Aird, K.M.; Zhang, G.; Li, H.; Tu, Z.; Bitler, B.G.; Garipov, A.; Wu, H.; Wei, Z.; Wagner, S.N.; Herlyn, M.; et al. Suppression of Nucleotide Metabolism Underlies the Establishment and Maintenance of Oncogene-Induced Senescence. Cell Rep. 2013, 3, 1252–1265.
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased Global Transcription Activity as a Mechanism of Replication Stress in Cancer. Nat. Commun. 2016, 7, 13087.
- Dominguez-Sola, D.; Ying, C.Y.; Grandori, C.; Ruggiero, L.; Chen, B.; Li, M.; Galloway, D.A.; Gu, W.; Gautier, J.; Dalla-Favera, R. Non-Transcriptional Control of DNA Replication by c-Myc. Nature 2007, 448, 445–451.
- Sankar, N.; Kadeppagari, R.-K.; Thimmapaya, B. C-Myc-Induced Aberrant DNA Synthesis and Activation of DNA Damage Response in P300 Knockdown Cells. J. Biol. Chem. 2009, 284, 15193–15205.
- Srinivasan, S.V.; Dominguez-Sola, D.; Wang, L.C.; Hyrien, O.; Gautier, J. Cdc45 Is a Critical Effector of Myc-Dependent DNA Replication Stress. Cell Rep. 2013, 3, 1629–1639.
- Maya-Mendoza, A.; Ostrakova, J.; Kosar, M.; Hall, A.; Duskova, P.; Mistrik, M.; Merchut-Maya, J.M.; Hodny, Z.; Bartkova, J.; Christensen, C.; et al. Myc and Ras Oncogenes Engage Different Energy Metabolism Programs and Evoke Distinct Patterns of Oxidative and DNA Replication Stress. Mol. Oncol. 2015, 9, 601–616.
- Bretones, G.; Delgado, M.D.; León, J. Myc and Cell Cycle Control. Biochim. Et Biophys. Acta (BBA)—Gene Regul. Mech. 2015, 1849, 506–516.
- Curti, L.; Campaner, S. MYC-Induced Replicative Stress: A Double-Edged Sword for Cancer Development and Treatment. Int. J. Mol. Sci. 2021, 22, 6168.
- Liou, G.-Y.; Storz, P. Reactive Oxygen Species in Cancer. Free Radic. Res. 2010, 44, 479–496.
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. C-Myc Can Induce DNA Damage, Increase Reactive Oxygen Species, and Mitigate P53 Function: A Mechanism for Oncogene-Induced Genetic Instability. Mol. Cell 2002, 9, 1031–1044.
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras Proteins Induce Senescence by Altering the Intracellular Levels of Reactive Oxygen Species. J. Biol. Chem. 1999, 274, 7936–7940.
- Dobbelstein, M.; Sørensen, C.S. Exploiting Replicative Stress to Treat Cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423.
- Cottini, F.; Hideshima, T.; Xu, C.; Sattler, M.; Dori, M.; Agnelli, L.; ten Hacken, E.; Bertilaccio, M.T.; Antonini, E.; Neri, A.; et al. Rescue of Hippo Coactivator YAP1 Triggers DNA Damage–Induced Apoptosis in Hematological Cancers. Nat. Med. 2014, 20, 599–606.
- Cottini, F.; Hideshima, T.; Suzuki, R.; Tai, Y.-T.; Bianchini, G.; Richardson, P.G.; Anderson, K.C.; Tonon, G. Synthetic Lethal Approaches Exploiting DNA Damage in Aggressive Myeloma. Cancer Discov. 2015, 5, 972–987.
- Herrero, A.B.; Gutiérrez, N.C. Targeting Ongoing DNA Damage in Multiple Myeloma: Effects of DNA Damage Response Inhibitors on Plasma Cell Survival. Front. Oncol. 2017, 7, 98.
- Herrero, A.B.; San Miguel, J.; Gutierrez, N.C. Deregulation of DNA Double-Strand Break Repair in Multiple Myeloma: Implications for Genome Stability. PLoS ONE 2015, 10, e0121581.
- Botrugno, O.A.; Bianchessi, S.; Zambroni, D.; Frenquelli, M.; Belloni, D.; Bongiovanni, L.; Girlanda, S.; Di Terlizzi, S.; Ferrarini, M.; Ferrero, E.; et al. ATR Addiction in Multiple Myeloma: Synthetic Lethal Approaches Exploiting Established Therapies. Haematologica 2020, 105, 2440–2447.
- Walters, D.K.; Wu, X.; Tschumper, R.C.; Arendt, B.K.; Huddleston, P.M.; Henderson, K.J.; Dispenzieri, A.; Jelinek, D.F. Evidence for Ongoing DNA Damage in Multiple Myeloma Cells as Revealed by Constitutive Phosphorylation of H2AX. Leukemia 2011, 25, 1344–1353.
- Dib, A.; Gabrea, A.; Glebov, O.K.; Bergsagel, P.L.; Kuehl, W.M. Characterization of MYC Translocations in Multiple Myeloma Cell Lines. J. Natl. Cancer Inst. Monogr. 2008, 2008, 25–31.
- Shou, Y.; Martelli, M.L.; Gabrea, A.; Qi, Y.; Brents, L.A.; Roschke, A.; Dewald, G.; Kirsch, I.R.; Bergsagel, P.L.; Kuehl, W.M. Diverse Karyotypic Abnormalities of the C-Myc Locus Associated with c-Myc Dysregulation and Tumor Progression in Multiple Myeloma. Proc. Natl. Acad. Sci. USA 2000, 97, 228–233.
- Chng, W.-J.; Huang, G.F.; Chung, T.H.; Ng, S.B.; Gonzalez-Paz, N.; Troska-Price, T.; Mulligan, G.; Chesi, M.; Bergsagel, P.L.; Fonseca, R. Clinical and Biological Implications of MYC Activation: A Common Difference between MGUS and Newly Diagnosed Multiple Myeloma. Leukemia 2011, 25, 1026–1035.
- Affer, M.; Chesi, M.; Chen, W.D.; Keats, J.J.; Demchenko, Y.N.; Tamizhmani, K.; Garbitt, V.M.; Riggs, D.L.; Brents, L.A.; Roschke, A.V.; et al. Promiscuous MYC Locus Rearrangements Hijack Enhancers but Mostly Super-Enhancers to Dysregulate MYC Expression in Multiple Myeloma. Leukemia 2014, 28, 1725–1735.
- Kuehl, W.M.; Bergsagel, P.L. MYC Addiction: A Potential Therapeutic Target in MM. Blood 2012, 120, 2351–2352.
- Misund, K.; Keane, N.; Stein, C.K.; Asmann, Y.W.; Day, G.; Welsh, S.; Van Wier, S.A.; Riggs, D.L.; Ahmann, G.; Chesi, M.; et al. MYC Dysregulation in the Progression of Multiple Myeloma. Leukemia 2020, 34, 322–326.
- Holien, T.; Våtsveen, T.K.; Hella, H.; Waage, A.; Sundan, A. Addiction to C-MYC in Multiple Myeloma. Blood 2012, 120, 2450–2453.
- Manier, S.; Salem, K.Z.; Park, J.; Landau, D.A.; Getz, G.; Ghobrial, I.M. Genomic Complexity of Multiple Myeloma and Its Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 100–113.
This entry is offline, you can click here to edit this entry!