Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
In humans, Interleukin-8 (IL-8 or CXCL8) is a granulocytic chemokine with multiple roles within the tumor microenvironment (TME), such as recruiting immunosuppressive cells to the tumor, increasing tumor angiogenesis, and promoting epithelial-to-mesenchymal transition (EMT).
- CXCL8-CXCR1/2 axis
- cancer
- tumor microenviroment
- immunotherapy
- interleukin-8
1. CXCL8-CXCR1/2 Signaling Pathway
The proinflammatory cytokine CXCL8 was initially found as a chemotactic agent for neutrophils in inflammatory diseases. Through both autocrine and paracrine signaling, the CXCL8-CXCR1/2 axis can recruit the neutrophil to clear bacteria and protect the host from infection. Due to the similarity of the pathogenesis of inflammatory diseases and cancer, more researchers have focused on the roles of the CXCL8-CXCR1/2 axis in cancer [22].
CXCL8 is a peptide with 72 amino acids and has a critical N-terminal motif of Glu-Leu-Arg (ELR). CXCL8 can be secreted by fibroblasts, CAFs, endothelial cells, epithelial cells, DCs, monocytes, macrophages, and cancer cells [23]. CXCL8 signals through two cell-surface receptors of CXCR1 and CXCR2. CXCR1 and CXCR2 are the G-protein-coupled receptor for a group of C-X-C chemokines. CXCR1 interacts with CXCL6 and CXCL8, whereas CXCR2 interacts with CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, CXCL7, and CXCL8. CXCL8-CXCR1/2 signaling with CAF, the microbiome, and immune cell help in recruiting granulocytes such as neutrophils and MDSCs to the site of the TME and contribute to tumor growth by enhancing angiogenesis and promoting cancer cell proliferation [24] and immune resistance [25] (Figure 1).
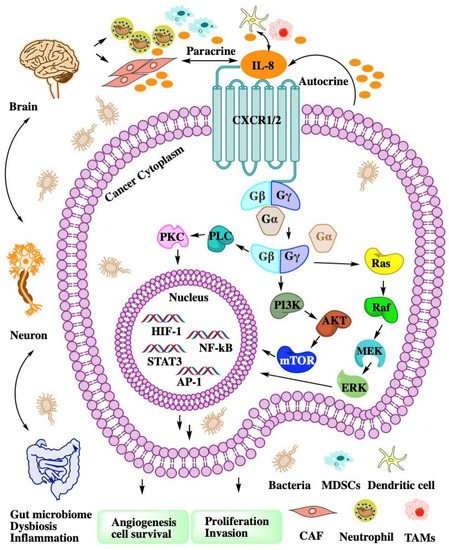
Figure 1. The intricate network of the CXCL8-CXCR1/2 axis in TME. CXCL8 binding to CXCR1/2 activates G-protein-mediated signaling cascades in cancer cell. CXCR1/2 activation leads to the dissociation of the Gα subunit from the Gβγ subunits. The signal of Gβγ subunits activate kinase to enhance angiogenesis, proliferation, and invasion. Cancer cell autocrine CXCL8 to recruit MDSCs or neutrophil to TME. Dysbiosis or inflammation affects myeloid cell recruitment through the gut–brain axis.
2. Interactions of the CXCL8-CXCR1/2 Axis and CAF
CAFs are activated fibroblast populations and the major cellular components of the TME in primary and metastatic cancers [26,27]. CAFs have different features from normal fibroblasts after infiltrating tumor tissue, such as enhanced proliferation and epigenetic changes to produce secreted factors [28]. CAFs are contributors to desmoplasia that can facilitate tumorigenicity, cancer cell proliferation, metastasis, and cancer immunotherapy resistance through complex interactions and intricate signaling [29] with other cell types in the TME [30].
The population of CAFs are highly heterogeneous because several progenitor cell types can be reprogrammed into CAFs [31]. Quiescent or resident fibroblasts are major progenitor cells of CAFs [32]. Hepatic [33] or pancreatic stellate cells [34] are the putative origin of CAFs. Adipocytes [35], endothelial cells [36], epithelial cells [37], and bone marrow cells [38] can be reprogrammed into CAFs. CAFs can also derive from multiple resident precursors, such as smooth muscle cells or mesenchymal stem cells [39]. Nevertheless, the precise origins of CAFs remain elusive because of the lack of lineage biomarkers.
CAFs have both tumor-promoting and tumor-suppressive functions [40]. The tumor-suppressive functions of CAFs [41] remain poorly understood. Part of the host defensive mechanism involves promotion of anticancer immunity, tumor-inhibitory signaling, tumor-restraining metabolism, and ECM-related physical barriers to tumor cell invasion and dissemination. Desmoplasia is the growth of fibrous or connective tissue in desmoplastic breast, lung, and pancreatic cancers. The desmoplastic reaction may form the desmoplasia or dense fibrosis around the tumor to restrict its growth and migration [42]. The mechanisms of tumor-promoting roles of CAFs are mainly regulatory functions via growth factors, cytokines, and chemokines contributing to angiogenesis; ECM remodeling; aberrant stroma, and an immunosuppressive TME [43]. Mass cytometry and single-cell analysis of pancreatic tumors and healthy pancreas samples found two stable and functionally distinct pancreatic fibroblast lineages. CD105-positive pancreatic fibroblasts promote tumor growth, but CD105-negative fibroblasts are highly tumor suppressive in a manner dependent on adaptive immunity [44].
CAF subsets have specialized secretory functions, such as cytokines, chemokines, and ECM molecules collagen I, which contribute to ECM remodeling and immunomodulatory function. Tumor–fibroblast interactions via soluble factors determine the final outcome of the tumorigenic process and affect the cancer therapy [45]. The CAF population is heterogeneous according to the cell origins, and the functional heterogeneity can be regulated by paracrine molecules such as CXCL8 and CXCR1/2 ligands [30]. CAFs can attract monocytes by secreting CXCL8 to enhance TAM enrichment and suppress NK cells’ function in colorectal cancer [46]. In gastric cancer tissues of chemoresistant patients, CXCL8 was highly expressed and located in CAFs by immunohistochemistry assay. A high serum CXCL8 level was associated with poor response to cisplatin therapy in gastric cancer patients [47]. CAFs are major sources of chemokines (CXCL1 and CXCL8) that recruit granulocytes (TAM and PMN-MDSC) to tumors. Combining a selective CSF1R inhibitor (JNJ-40346527) with a CXCR2 antagonist (SB225002) blocked granulocyte recruitment and demonstrated a strong antitumor effect, which was further improved by the addition of antiprogrammed cell death protein 1 (PD-1) [48].
Chronic inflammation and proinflammatory cytokines tumor necrosis factor α (TNFα) and interleukin 1β (IL-1β) can induce the conversion of MSCs to inflammatory CAFs. These CAFs secrete prometastatic chemokines including CXCL6 and CXCL8 in Luminal-A breast cancer cells and enhance migration [49]. CXCL8 can induce normal ovarian fibroblasts to CAFs and stimulate xenograft tumor growth in mice. The ovarian cancer cell stemness was promoted by the CXCL8 secreted from CAFs through the Notch3 signaling pathway [50]. Gastric cancer extracellular vesicles transfer various miRNAs and induce chemokines such as CXCL1 and CXCL8 expression in CAFs. Aberrant chemokine CXCL1 and CXCL8 expression in CAFs was closely associated with tumor progression and poorer survival in gastric cancer patients [51]. CAFs demonstrate a high level of basal secretory autophagy in head and neck squamous cell carcinoma. Secretory autophagy is involved in the export of cellular inflammatory mediators such as IL-6 and CXCL8. Combination therapy using autophagy inhibition with cisplatin significantly reduced tumor volume [52].
Stromal cells such as CAFs and MSCs enhance the triple-negative subtype of breast cancer metastasis-related phenotypes, including angiogenesis, migratory, and invasive properties, by releasing inflammatory chemokines such as CCL2 and CXCL8 [53]. Notch 1 activation is required for the induction of CXCL8 in tumor–stroma interactions and consequently for prometastatic activities [54]. Androgen receptor signaling in CAFs affects prostate cancer cell migration mediated by CXCL8 and CCL2 [55]. CAFs express CXCR2 and respond to paracrine signals of pancreatic cancer cells by upregulating CXCR2 ligands such as CXCL1, CXCL7, and CXCL8. CXCR2 knockout in a pancreatic ductal adenocarcinoma syngeneic mouse model suppressed angiogenesis and induced an antitumor response, but increased CAF activation, fibrosis, and metastasis in a mutation-dependent manner [56].
The CAF response to chemotherapy is highly variable and may impact on cancer therapy outcome [57]. Traditional maximum-tolerated dose chemotherapy induces CAF activation and results in the expression and secretion of ELR motif-positive chemokines such as CXCL6 and CXCL8 [58]. These chemokines signal though CXCR2 on cancer cells to transform into tumor-initiating cells, thus promoting aggression and treatment resistance. While the same overall dose administered as a low-dose metronomic chemotherapy largely prevented CAF activation and enhanced treatment response, it improved survival in mice [58]. CAF-secreted IL-6 and CXCL8 induce Bromodomain-containing protein 4 (BRD4) protein expression and lead to chromatin remodeling and Bromodomain and Extraterminal (BET) inhibitor resistance in colorectal cancer (CRC). Inhibition of IL-6/CXCL8-JAK2 signaling sensitized BET inhibitors in a CRC mouse xenograft model [59]. Simultaneous blocking of IL-6 and CXCL8 can inhibit CAF-induced human melanoma cell invasiveness using neutralizing antibodies in a 3D spheroid invasion assay [60]. Senescent human fibroblasts can secrete IL-6 and CXCL8 to promote cancer cell invasion and metastasis [61]. Senescent CAFs are a pathologically relevant fibroblast population that secrete excess CXCL8 to promote pancreatic cancer invasion [62]. A new subset of CD10+GPR77+CAFs induce cancer stem cells’ (CSCs) enrichment and chemoresistance by secreting IL-6 and CXCL8 in cancer. CD10+GPR77+CAFs constitute a supporting niche for CSCs, and its high expression correlates with poor survival in breast and lung cancer patients [63].
3. Therapeutic Targeting of the CXCL8-CXCR1/2 Axis in Cancer
Chemokine CXCL8 activates both CXCR1 and CXCR2. All of them have been shown to be upregulated in acute or chronic inflammatory diseases including cancer. CXCR1 and CXCR2 constitute the primary mechanism for the recruitment of neutrophils and MDSCs, which could enhance tumor progression and suppress immune therapy efficacy. Blocking the CXCL8-CXCR1/2 axis with small molecules (Figure 2) or antibodies should be a promising therapeutic strategy to overcome immune suppression in the TME.
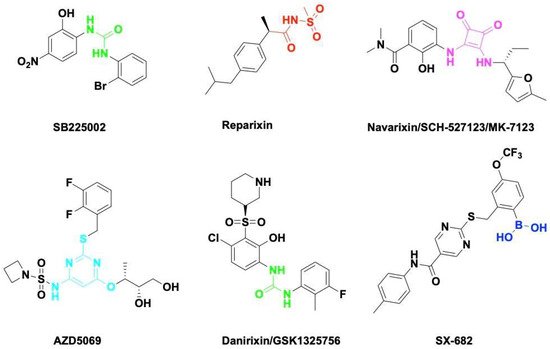
Figure 2. Small molecule antagonists targeting CXCR1/2. Blocking CXCR1 or CXCR2 impairs immune suppressive cell recruitment and angiogenesis to enhance cancer therapy efficacy.
SB225002 was first reported as a small molecule CXCL8 inhibitor binding to CXCR2. SB225002 selectively blocked CXCL8-induced neutrophil chemotaxis and margination in rabbits [164]. Blocking CXCR2 with SB225002 has proved to inhibit tumor progression in breast cancer [165], ovarian cancer [166], acute myeloid leukemia [167], and nasopharyngeal carcinoma [168]. Reparixin is a non-competitive allosteric inhibitor of CXCR1 and CXCR2 that could inhibit polymorphonuclear cell recruitment in vivo. The activity of Reparixin on CXCR1 was 100-fold higher than on CXCR2 [169]. CXCR1 has been identified as a druggable target for breast cancer stem cells [170].
Navarixin is an orally selective, CXCR1 and CXCR2 receptor antagonist that impairs neutrophil recruitment in rodents and monkeys [171]. Navarixin decreased tumor cell proliferation and angiogenesis in a melanoma mouse model [172]. A phase II clinical trial (NCT03473925) assessed the efficacy and safty of navarixin in combination with anti-PD1 pembrolizumab in NSCLC, Castration-Resistant Prostate Cancer and Colorectal Cancer (Table 1). AZD5069 is a reversible CXCR2 antagonist that inhibits CXCL8 binding and neutrophil chemotaxis [173]. AZD5069 treatment inhibits TAM infiltration and vessel formation, increases CD4+/CD8+ T-cell infiltration, and suppresses tumor growth in advanced prostate cancer [174]. Combination therapy of AZD5069 with anti-PD1 Durvalumab (MEDI4736) was studied in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck (NCT02499328) and metastatic pancreatic ductal adenocarcinoma (NCT02583477). Danirixin is also a reversible and selective antagonist of CXCR2 that may be of benefit in diseases of excess neutrophilia [175]. The clinical trial of danirixin in healthy, elderly human volunteers demonstrated that it was safe and tolerable, and no serious adverse events were reported [176,177]. A recent study showed that danirixin suppressed breast cancer migration, invasion, and metastasis mediated by CXCL8 and TAMs [178].
Table 1. Selected clinical trials targeted in CXCL8-CXCR1/2 axis.
| Drug (Manufacturer) | Target | Therapeutic Combinations | Cancer Type | Phase | Clinical Trials |
Recruitment Status |
|---|---|---|---|---|---|---|
| SX-682 (Syntrix Biosystems, Inc.) | CXCR1/2 | Pembrolizumab (anti-PD-1) | Metastatic Melanoma | Phase I | NCT03161431 | Recruiting |
| Nivolumab (anti-PD-1) | Metastatic Colorectal Carcinoma | Phase Ib/II | NCT04599140 | Recruiting | ||
| Nivolumab (anti-PD-1) | Pancreatic Cancer | Phase I | NCT04477343 | Recruiting | ||
| Bintrafusp alfa (anti-PD-L1/TGF-β) CV301 (cancer vaccine) |
Advanced Solid Tumors | Phase I | NCT04574583 | Active, not recruiting | ||
| AZD5069 (AstraZeneca) | CXCR2 | Durvalumab (anti-PD-L1) | Advanced Solid Tumors | Phase Ib/II | NCT02499328 | Active, not recruiting |
| Durvalumab (anti-PD-L1) | Metastatic Pancreatic Ductal Adenocarcinoma | Phase II | NCT02583477 | Completed | ||
| Navarixin (Merck Sharp & Dohme Corp.) | CXCR1/2 | Pembrolizumab (anti-PD-1) | Advanced Solid Tumors | Phase II | NCT03473925 | Completed |
| HuMax-IL8 (Bristol-Myers Squiibb) | CXCL8 | Nivolumab (anti-PD-1) | Head and Neck Squamous Cell Carcinoma | Phase II | NCT04848116 | Recruiting |
| Nivolumab (anti-PD-1) | Prostate Cancer | Phase Ib/II | NCT03689699 | Recruiting | ||
| Nivolumab (anti-PD-1) | Pancreatic Cancer | Phase II | NCT02451982 | Recruiting | ||
| Nivolumab (anti-PD-1) | Hepatocellular Carcinoma | Phase II | NCT04050462 | Recruiting | ||
| Nivolumab (anti-PD-1) | Advanced Cancers | Phase I/II | NCT03400332 | Recruiting | ||
| Nivolumab (anti-PD-1) | Non-small Cell Lung Cancer | Phase II | NCT04123379 | Recruiting |
SX-682 is a novel CXCR1/2 chemokine receptor boronic acid antagonist with potential anticancer activities. Orally bioavailable SX-682 enhanced NK cell activation and therapeutic efficacy by inhibiting MDSC accumulation in the TME [93]. A combination of SX-682 with anti-PD1 caused a reduction in tumor burden b7 increasing CD8+ T-cell infiltration and decreasing neutrophil accumulation in non-small cell lung cancer [179]. Treatment of tumor-bearing mice with SX682 reduced intertumoral MDSCs, increased CD8+ T-cell recruitment, and inhibited tumor growth in melanoma and breast cancer [180]. Several clinical trials were designed to assess the efficacy of SX-682 in combination with anti-PD1 Nivolumab or Pembrolizumab in metastatic melanoma, colorectal carcinoma, and pancreatic cancer (Table 1). Fully humanized neutralizing antibody ABX-IL8 inhibits angiogenesis, tumor growth, and metastasis in melanoma [181] and bladder cancer [182] through downregulation of MMP-2. HuMax-IL8(BMS-986253) is another fully human anti-CXCL8 monoclonal antibody. A phase I clinical trial of HuMax-IL8 (NCT02536469) showed no objective tumor responses, but it is safe and well tolerated [183]. Phase II clinical trials of HuMax-IL8 plus anti-PD1 Nivolumab are ongoing in patients with advanced solid tumors (Table 1).
Chimeric antigen receptor (CAR) T-cell therapy has shown clinical efficacy for hematological malignancies but still remains a challenge for solid tumors [184]. The major obstacles of CAR-T therapy in solid tumors are tumor heterogeneity, an immunosuppressive TME, and T-cell trafficking/infiltrating to the tumor. CXCR1/2-modified CAR-T cells enhance T-cell trafficking, persistence of T cells in the tumor, long-lasting immunologic memory, and therapy efficacy in aggressive solid tumors such as glioblastoma, ovarian, and pancreatic cancer [185].
4. CXCL8 as a Prognosis Biomarker in Cancer Therapy
Tumor burden (or tumor size) predicts response to immunotherapy in patients with cancer. A large TME and a small TME are characterized by different cell populations and responses to specific interventions [186]. Radiologic imaging such as computed tomographhy (CT) is the most commonly used technology for tumor burden monitoring even though it has limitations for the evaluation of the response to immunotherapy. Functional imaging techniques [187] such as positron emission tomography (PET) or electron paramagnetic resonance (EPR) can sense the TME hypoxia and visualize tissue redox status non-invasively, which assists image-guided diagnoses and efficacy evaluations of cancer therapy [188]. CXCL8 serum concentrations can accurately reflect the tumor burden of patients following antitumor therapy and have prognostic significance [189].
CXCL8 could be a serum biomarker by which to predict clinical benefit from immune checkpoint blockade in melanoma and NSCLC patients. Serum CXCL8 levels significantly decreased in responding patients treated with anti-PD1 nivolumab or pembrolizumab, which were associated with longer overall survival [190]. In gemcitabine-refractory patients with pancreatic cancer, plasma CXCL8 is a useful circulating biomarker for predicting resistance to nanoliposomal irinotecan therapy [191]. Baseline IL-6/CXCL8 can predict objective response and overall survival in patients with advanced HCC treated with sorafenib [192].
This entry is adapted from the peer-reviewed paper 10.3390/molecules27010137
This entry is offline, you can click here to edit this entry!