Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Cholinergic receptors are activated by acetylcholine, a neurotransmitter that is released by the motor neurons for sensory and motor processing.
- cholinergic receptors
- cholinergic neurons
- acetyl cholinesterase
1. Cholinergic Receptors
Cholinergic receptors are activated by acetylcholine, a neurotransmitter that is released by the motor neurons for sensory and motor processing. The cholinergic system includes nicotinic acetylcholine receptors (nAChRs) and muscarinic acetylcholine receptors (mAChRs) which are mainly responsible for signal transduction of autonomic and somatic nervous system. Nicotinic receptors function as ionotropic ligand-gated receptors and are receptive towards the agonist nicotine, whereas muscarinic receptors function as G-protein coupled receptors and are receptive towards muscarine [1]. The cholinergic neural transmission modulates memory and is associated with normal and abnormal cognitive functioning [2]. Thus, memory impairment and cognitive decline related to aging and dementia are associated with cholinergic system dysfunction [3]. Drugs acting on the cholinergic network may represent an exciting therapeutic avenue for the treatment of AD.
AD pathology is characterized by the loss of cholinergic neurons and the gradual down regulation of the brains acetylcholine level, which is influenced by excess acetylcholinesterase (AChE) [4]. AChE, a serine enzyme found at neuromuscular joints and cholinergic synapses, hydrolyzes ACh to acetate and choline. The hydrolysis action terminates the impulse transmission at cholinergic synapses [5]. Acetylcholinesterase inhibitors (AChEi) treat AD symptoms by improving cholinergic neurotransmission; however, acetylcholine esterase inhibitors only delay cognitive decline by increasing the level of acetylcholine in the brain. It does not change AD’s underlying pathology regarding continued loss of cognitive function [6][7].
In the search for new medicines to treat cognition and memory, researchers continuously look for molecules and compounds which can target acetyl cholinesterase (AChE). Derived from various flavones, isoflavones, flavanols, anthocyanidins, curcuminoids and stilbenes these compounds and play an important role in inhibiting the AChE enzyme [8]. The key molecules found in the cholinergic system are shown in Figure 1.
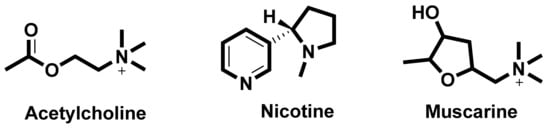
Figure 1. Key molecules comprising the cholinergic system.
2. Nicotinic Acetylcholine Receptors (nAChRs)
Nicotinic acetylcholine receptors are cholinergic receptors that respond to acetylcholine and bind to nicotine. They represent the heterogeneous family of ionotropic receptors which mediate neurotransmission through ligand-gated ion channels [9]. The nicotinic AChRs are located in muscles, the central nervous system (CNS) and peripheral nervous system (PNS). They are responsible for neuromuscular transmission that causes muscular contraction at skeletal neuromuscular junction and are also involved in synaptic transmission in the CNS and PNS [10]. In addition, nAChRs also influence cognition and memory function in the brain and control the pre-synaptic release of neurotransmitters such as dopamine [11]. These receptors are large pentameric structures with a molecular mass of ~290 KD. Each receptor consists of five types of protein subunits. These subunits can either be homologous (identical subunits) or heterologous (different subunits) [12].
The nAChRs, in human beings are arranged in 16 homomeric or heteromeric subunits, consisting of a diverse set of complex subtypes such as, α1–7, α9–10, β1–4, γ, δ and ε substructures. Among these substructures, α7 and α4β2 subunits are overly expressed in the CNS. Subtype α4β2 subunits have high affinity for nicotine and cytosine and a lower affinity for α-BTX (α-bungarotoxin), whereas, α7 subunit has a high affinity for α-BTX and lower affinity for nicotine and cytosine. The subtype α7 has five subunits and is crucial in AD pathology because these subunits are involved in memory and learning functions and participates in cholinergic anti-inflammatory pathways in relation to the autoimmune disorders. The five α7 subunits are homologous receptors and are commonly referred as α7 nAChRs. It is the only α-BTX receptor identified in mammalian brain. These receptors have an N–terminal peptide and a ligand binding site that has a high affinity for α-BTX and its agonists [13].
Subtype α7 nicotinic AChRs in hippocampal astrocytes can regulate the calcium signaling cascade in the CNS and contribute to cholinergic signaling. These receptors exhibit a high permeability to calcium compared to the sodium ion. On activation, these receptors produce a calcium transient in the cell and increase the intracellular concentration of free calcium. The process takes place via calcium induced calcium release and is triggered by the voltage gated calcium channels. The calcium signaling cascade in astrocyte is different compared to the activation of these receptors on the neurons [14]. In neurons, the activated presynaptic α7nAChR increases the flux of Ca2+, depolarizes the presynaptic membrane and merges this presynaptic membrane with vesicles containing neurotransmitters in the synapse. As a result, this leads to exocytosis and induces the release of neurotransmitters, such as glutamic acid, norepinephrine (NE), ACh, dopamine (DA) and γ-amino butyric acid (GABA).
Nicotinic AChRs have important role in regulating the release of pro-inflammatory cytokines in macrophages, brain astrocytes and microglia [15]. These receptors mediate the cholinergic signaling, especially in learning and memory function. In particular, α7-nAChR modulates the excitatory neurotransmitter release, improves learning and memory ability and enhances the cognitive function. It has been reported that in the brain of AD animal models and AD patients, the functioning level of α7nAChRs along with the level of expression is altered [16]. Higher level of α7nAChR is observed in the brain of early embryonic stage and this level progressively changes with age, inferring the importance of α7nAChR in growth, development, and aging [5]. Thus, α7nAChR may participate in AD pathogenesis and may serve as a novel therapeutic target for AD treatment.
2.1. Interaction between Amyloid Beta and α7nAChRs
The subunit α7nAChR is involved in cholinergic signaling and has been associated with amyloid beta deposition and AD pathogenesis. Relevant to cognition, synaptic plasticity and the neurotoxic properties of Aβ peptides, several studies have proposed both agonistic and antagonistic relationships between Aβ and α7nAChRs. Activation or inhibition of α7nAChRs blocks Aβ peptide mediated neuronal cell death and is considered a critical step in the identification of potential therapeutic strategies to treat AD pathogenesis. Accumulated evidence suggests that subtype α7 nicotinic acetylcholine receptors mediate Aβ induced neurotoxicity in hippocampal neurons [17] and Aβ induced tau phosphorylation via activation of tau kinases, extracellular-signal-regulated kinase (ERK), and c-Jun N-terminal kinase (JNK-1) [18]. In these cases, α7nAChRs demonstrates an agonistic effect on Aβ peptide induced neuronal cell death. Inhibition of the α7nAChR subunit, rather than activation, may imply a pathway that eliminates the neurotoxic properties from Aβ peptides. Aβ induced neurotoxicity is mediated by the up regulation of α7nAChRs in hippocampal neurons. Liu et al. emphasized that α7nAChRs up regulation produces Aβ induced neuronal hyperexcitation and possibly, AD pathogenesis [19]. The up regulated α7nAChRs are also responsible for the down regulation of ERK2 mitogen-activated protein kinase (MAPK) activity that is critical in hippocampus synaptic plasticity and learning. The in vitro and in vivo studies demonstrate that Aβ42 pairs up with MAPK cascade via α7nAChRs, leading to the up regulation of α7nAChR. The ERK MAPK cascade may regulate the production of Aβ42 peptides and the down regulated ERK2 MAPK may alleviate the accumulation of Aβ42 peptides [20].
Immunohistochemical studies of human brain tissues confirm the agonistic relationship between Aβ peptides and α7nAChRs. Wang et al., illustrated that Aβ 1-42 has high affinity with α7nAChR and binds to form a stable complex that inhibits the release of ACh and alters Ca2+ homeostasis in the cholinergic neuron causing neuronal dysfunction (Figure 2). They have also reported that Aβ1-42 mediates the death of human neuroblastoma cells that overexpress α7 nicotinic acetylcholine receptors, which contrast with α7nAChR agonists, nicotine and epibatidine that protect human neuroblastoma cells from neuronal death induced by amyloid beta peptides [21]. Likewise, in vitro studies have also shown that nicotine inhibits the formation of Aβ fibril from Aβ1-42 and disrupts preformed Aβ fibrils protecting the neurons from Aβ toxicity via the up regulation of nicotinic receptors [22]. Similar studies have also provided evidence demonstrating nicotine’s effectiveness at reducing the insoluble Aβ peptide plaques in treated transgenic mouse brain of [23]. Together, these studies suggest that the reduction of amyloid beta peptide in brain may be mediated by α7nAChRs and nicotinic drug may prove a novel protective AD therapy.
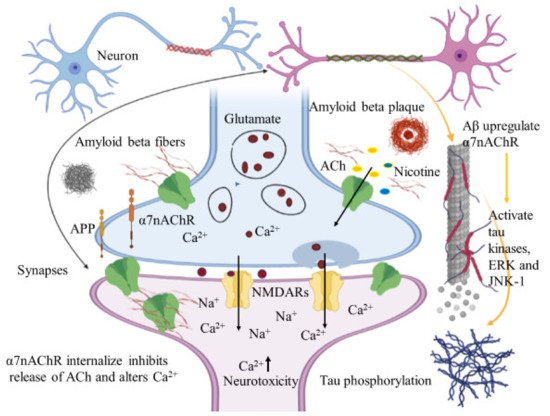
Figure 2. Amyloid beta interacts in a futile cycle with α7nAChRs. Amyloid beta interacts with α7nAChRs increasing the flux of intracellular Ca2+ in the presynaptic region. Ca2+ releases glutamate, resulting in postsynaptic depolarization followed by NMDAR activation. Long-term activation desensitizes and internalizes NMDARs and α7nAChRs post synaptically, inhibits the release of ACh, and alters Ca2+. In addition, Aβ up regulates α7nAChRs and induces tau phosphorylation via the activation of tau kinases, ERK, and JNK-1. The figure was created using tools obtained from BioRender.com.
The cerebral cortex of AD patients has low AChE levels within the amyloid plaques [24]. The activity of AChE increases around the amyloid plaques and is believed to be caused by the direct action of Aβ on AChE [25]. AChE is a cholinergic enzyme that terminates the synaptic transmission between the synapses [26]. Fodero et al. have demonstrated that the effect of Aβ1-42 on AChE is due to the agonist effect of Aβ on α7nAChRs. In primary cortical neurons, α7nAChRs mediate the Aβ1-42 induced AChE increase. The inhibitors of α7nAChRs and L- or N- type voltage dependent calcium channels (VDCCs) block the effect of Aβ on AChE and α7nAChR agonist s increases the level of AChE [27]. In this case, AChE inhibitors have been used to reduce ACh breakdown and enhance their levels. This subsequently makes the neurotransmitter available to the nicotinic and muscarinic receptors, enabling them to increase cholinergic signaling and thereby reducing AD memory deficits [28].
AD is pathologically characterized by the presence of extracellular amyloid plaques and intracellular NFTs. The α7nAChR subtype is involved in synaptic plasticity in the brain and Aβ1-42 is closely associated with AD pathogenesis. Such agonistic results suggest that Aβ may exert some of its toxicity through α7nAChRs and thus provide a possible pathway for AD treatment by blocking the action of Aβ 42 on α7nAChRs. In contrast, several other reports have elucidated the antagonistic relationship between α7nAChRs and Aβ peptide. The nicotinic acetylcholine receptors with the subunit α7 mediate synaptic current and plasticity in the brain. They modulate cellular function in the nervous system and their responses affect cognitive processes and memory forming abilities. However, the responses of α7 containing nicotinic receptors are blocked by the presence of nanomolar concentrations of Aβ1-42 in the hippocampal neurons, thereby affecting their proposed cognitive roles and leading to learning and memory dysfunction [29]. In a different study, Puzzo et al. demonstrated that a picomolar concentration of Aβ, exerted a positive modulatory effect on hippocampal synaptic plasticity and memory, whereas a higher concentration resulted in neuronal dysfunction and cognitive failure [30]. Nonetheless, these studies suggest that nicotinic receptors are a subject of interest for treating AD pathogenesis. Their central role in synaptic regulation offers an exciting opportunity for therapeutic development that specifically targets these receptors or alternatively intercepts the action of Aβ peptides on the nicotinic receptors.
2.2. Allosteric Modulation of nAChRs
In addition to the hallmark Aβ plaques and hyper phosphorylated tau proteins, neuroinflammation appears to have a significant role in the development and progression of AD pathogenesis. The neuro-inflammatory process is marked by microglial activation and is considered to be induced by the binding of Aβ peptides to cluster of differentiation 36 (CD36) protein and toll like receptors (TLR) of four and six heterodimers, such as TLR4 and TLR6 [31]. Activation eventually leads to the production of proinflammatory cytokines, for example, interleukins (IL-1β, IL-6, IL-8) and tumor necrosis factor (TNF), along with anti-inflammatory cytokines, such as transforming growth factor β (TGFβ), chemokine, and small messenger molecules, namely, nitric oxide and reactive oxygen species, which over time, cause nerve impairment and neuronal death [32].
In this context, the microglial activation leading to neuroinflammation has also been reported to take place via the brain cholinergic pathways involving α7 nAChRs, where the release of microglial TNF-α, as induced by lipopolysaccharide (LPS) through activation of α7 nAChRs, is regulated by the acetylcholine and nicotine levels [33]. The use of nAChR allosteric modulators is a promising strategy to reduce neuroinflammation [34] earlier. The current treatment, which uses nAChR agonist and AChE inhibitors, is limited by their desensitization properties [35].
In AD, allosteric modulation intensifies the ACh activity at pre- and post-synaptic nAChR. This is particularly important because the presynaptic ACh level is significantly lower and impaired in AD patients. Allosteric modulators of nAChRs increases the presynaptic ACh level and enhances the cholinergic nicotinic neurotransmission by amplifying the interaction between the nAChR and ACh. This presynaptic release of ACh is considered to be mediated by α7 and α4β2 nAChR [36]. Accordingly, some of the allosteric modulators of nAChRs are capable of producing positive modulatory effects on nAChR. These positive allosteric modulators (PAMs) may have therapeutic potential. For instance, desformylfulstrabromide (dFBr), PAM of α4β2 receptors, enhances cognition in rat [37]. Galantamine, type I PAM for α7 and α4β2 nAChR, elevates ACh levels and ameliorates cognition in AD patients [38], and cotinine, PAM of α7 receptor, is neuroprotective, improves memory in primates, and lowers the Aβ burden in AD mice [39].
3. Muscarinic Acetylcholine Receptors
The cholinergic receptors most responsive to muscarine are referred to as the muscarinic acetylcholine receptors (mAChRs). Members of the metabotropic receptor class, mAChRs mediate the physiological action of acetylcholine in the CNS and PNS using G-protein for as the signaling mechanism [40]. Found throughout the human body (e.g., renal and cardiovascular systems, gastrointestinal tract, eyes, brain, salivary glands) mAChRs are responsible for many distinct physiological functions including motor control, cognition, and sensory processing, dependent upon location and receptor subtype [41][42]. The mAChRs are the receptive group of the acetylcholine neurotransmitter and mediate cholinergic transmission related to learning, memory, and cognition in the forebrain region thus the implication in AD [43].
Muscarinic receptors are part of the ligand-gated G-protein coupled receptor and perform either as a simulative regulative G-protein (Gs) or an inhibitory regulative G-protein (Gi) [44]. The muscarinic AChRs have been divided into five receptor subtypes, M1-M5. Subtypes M1, M2 and M4 muscarinic receptors are adequately present in the brain, whereas M3 and M5, are sparsely distributed [45][46]. The receptors M1, M3, and M5 are stimulatory receptors that interact with the Gq/11 G protein family and are involved in the stimulation of phospholipase C and instigation of the phosphatidylinositol trisphosphate cascade, resulting in the intracellular Ca2+ mobilization and the activation of protein kinase C. In contrast, the inhibitory receptors M2 and M4, interact with the Go/i family of G proteins. This set of receptors are responsible for inhibiting adenylyl cyclase i and activating G protein-gated potassium channels. The inhibitory action of adenylyl cyclase reduces the level of protein kinase A, ultimately decreasing cAMP (cyclic adenosine monophosphate) levels in the cell [47].
The subtype M1 muscarinic receptor extends throughout the brain and is found primarily in the hippocampus, cerebral cortex, amygdala, and corpus striatum [47]. Amyloid plaques and NFTs develop in the cerebral cortex and hippocampus Given that M1 receptors are highly expressed in these sections, the M1 muscarinic receptor is a potential therapeutic target for restoring cholinergic signaling [48]. M1 muscarinic receptors are associated with numerous physiological functions, including neuron excitation, synaptic plasticity, learning and hippocampal-based memory, neuronal differentiation during early development, modulation of cognition, and short-term memory [49]. Besides the obvious AD associated plaques and tangles, presynaptic cholinergic hypo function is also considered an equally attributive factor in progressive dysfunctional cognition. Lending weight to the cholinergic hypothesis, post-synaptic M1 mAChR remains unaffected, and in this case, several studies have suggested M1 muscarinic agonists as prospective AD treatment candidates [50].
M1 muscarinic agonists have been reported to reduce amyloid-β peptides and tau pathologies in the hippocampus and cortex regions of an AD mouse model. Caccamo et al. administered AF267B, a low molecular weight M1 muscarinic agonistic, in the 3xTg-AD model of Alzheimer disease. 3xTg-AD model mice experienced a reduction in both Aβ and tangles in the hippocampus and cortex. Cognitive deficits related to spatial tasks were additionally reversed. Likewise, they also reported that dicyclomine, M1 antagonist, amplified Aβ and tau pathologies, highlighting the importance of M1 agonist against the biomarkers of AD pathologies. M1 activators modulated the amyloid precursor protein (APP) processing away from the amyloidogenic pathway by generating α-secretase products via the activation of protein kinase C (PKC) and extracellular regulated kinase (ERK ½), ultimately diminishing the Aβ peptides. The activation of PKC also reduced the activity of tau kinase, glycogen synthase kinase (GSK) 3β, decreasing tau pathologies in the AD animal model [48]. The M1-selective agonist, VU0364572, was administered to 5XFAD mice, by another group, which showed that levels of soluble and insoluble Aβ40, 42 were significantly reduced in the hippocampus and cortex of the animal preventing memory impairment as demonstrated by the Morris water maze task [51]. These findings reinforce the development of M1 activators as a disease-modifying treatment for AD pathology. The activation of PKC is linked with increased APP metabolism, secretion, and processing [52]; selective cholinergic agonists and M1 muscarinic agonists increase PKC activity and serve as an effective AD modifying therapy [53]. M1 mAChR’s significance to APP processing and amyloid pathology has also been demonstrated in vitro and in vivo AD transgenic mouse model studies. Removal of the receptor increased amyloid pathology, indicating that M1 mAChR is instrumental in modulating amyloidogenic APP processing in neurons with M1 activators as potential therapeutic resources for treating AD [54].
Like M1 agonists, M2 antagonists are also considered a prospective therapeutic measure for treating AD behavioral and cognitive symptoms. As mentioned previously, increased M2 mAChR activity decreases the adenylyl cyclase activity, which later decreases cAMP levels, ultimately leading to memory loss as well as reduce synaptic plasticity. Decreased cAMP levels correlate with depression and dementia severity. It has been reported that blocking M2 mAChR increases the level of ACh in the brain. The elevation of ACh then activates the postsynaptic M1 mAChR, which is involved in cholinergic signaling, eventually improving the cognitive processes [55].
Acetylcholinesterase inhibitors (AChE-Is), such as donepezil, galantamine, tacrine, and rivastigmine have been used for the symptomatic treatment of AD to ameliorate the cognitive function status. These inhibitors do not modify the disease progression, but rather they inhibit the action of the AChE enzyme on ACh and increase the ACh level to bind with AChRs to improve cholinergic signaling [56]. However, this treatment lasts for shorter period of time and is not considered effective. M1 muscarinic agonists offer a greater advantage than AChE-Is and can activate cholinergic receptors, which is less susceptible to ACh degeneration of. The successful development of M1 selective agonists is essential to treating AD pathology.
Various studies have demonstrated that muscarinic and nicotinic agonists are viable therapeutic targets for the treatment of AD. Several preclinical and clinical phases studies have shown that the agonists viz. xanomeline, encenicline, nelonicline (ABT-126), AF102B effectively treat mild to moderate stages of AD through neural regeneration and decreasing amyloid-β concentration [57]. Most of these agonists mimic the action of the physiological neurotransmitter (ACh) found in the CNS. Although these agonists are possible therapeutic targets, and passed initial clinical trials, they were suspended or terminated during phase three of their respective clinical trials. Reasons for failure included cross-activity with other receptors, especially 5-HT3 and insufficient selectivity with cholinergic receptors. Rather than developing a thorough understanding of AD mechanisms and recruiting diverse populations for clinical studies, most of the agonists were designed solely to decrease amyloid-β concentration Most of the agonists were designed solely to decrease. Many laboratories remain focused on cholinergic receptor agonists by manipulating or modifying existing drugs or by designing completely new moiety using computational drug design tools. Enrolling patients during the earliest stages of the disease, selecting more diverse participants, and paying attention to specific structure-based and ligand-based drug design could increase AD treatment therapeutic target success.
References
- Carlson, A.B.; Kraus, G.P. Physiology, Cholinergic Receptors. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK526134/ (accessed on 6 September 2020).
- Maurer, S.V.; Williams, L. The Cholinergic System Modulates Memory and Hippocampal Plasticity via Its Interactions with Non-Neuronal Cells. Front. Immunol. 2017, 8, 1489.
- Bertrand, D.; Wallace, T.L. A Review of the Cholinergic System and Therapeutic Approaches to Treat Brain Disorders. Curr. Top. Behav. Neurosci. 2020, 45, 1–28.
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115.
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335.
- Akıncıoğlu, H.; Gülçin, İ. Potent acetylcholinesterase inhibitors: Potential drugs for Alzheimer’s disease. Mini-Rev. Med. Chem. 2020, 20, 703–715.
- Rosengarten, B.; Paulsen, S.; Burr, O.; Kaps, M. Neurovascular coupling in Alzheimer patients: Effect of acetylcholine-esterase inhibitors. Neurobiol. Aging 2009, 30, 1918–1923.
- D’Onofrio, G.; Sancarlo, D.; Ruan, Q.; Yu, Z.; Panza, F.; Daniele, A.; Greco, A.; Seripa, D. Phytochemicals in the treatment of alzheimer’s disease: A systematic review. Curr. Drug Targets 2017, 18, 1487–1489.
- Karlin, A.; Akabas, M.H. Toward a structural basis for the function of nicotinic Acetylcholine receptors and their cousins. Neuron 1995, 15, 1231–1244.
- Hogg, R.C.; Raggenbass, M.; Bertrand, D. Nicotinic acetylcholine receptors: From structure to brain function. Rev. Physiol. Biochem. Pharmacol. 2003, 147, 1–46.
- Lykhmus, O.; Voytenko, L.; Koval, L. α7 Nicotinic acetylcholine receptor-specific antibody induces inflammation and amyloid β42 accumulation in the mouse brain to impair memory. PLoS ONE 2015, 10, e0122706.
- Mitra, S.; Khatri, S.N.; Maulik, M.; Bult-Ito, A.; Schulte, M. Allosterism of Nicotinic Acetylcholine Receptors: Therapeutic Potential for Neuroinflammation Underlying Brain Trauma and Degenerative Disorders. Int. J. Mol. Sci. 2020, 21, 4918.
- Ma, K.G.; Qian, Y.H. Alpha 7 nicotinic acetylcholine receptor and its effects on Alzheimer’s disease. Neuropeptides 2019, 73, 96–106.
- Sharma, G.; Vijayaraghavan, S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc. Natl. Acad. Sci. USA 2001, 98, 4148–4153.
- De Jonge, W.J.; Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 2007, 151, 915–929.
- Shen, J.; Wu, J. Nicotinic cholinergic mechanisms in Alzheimer’s disease. Int. Rev. Neurobiol. 2015, 124, 275–292.
- Liu, Q.; Xie, X.; Emadi, S.; Sierks, M.R.; Wu, J. A novel nicotinic mechanism underlies beta-amyloid induced neurotoxicity. Neuropharmacology 2015, 97, 457–463.
- Wang, H.Y.; Li, W.; Benedetti, N.J.; Li, D.H.S. Alpha 7 nicotinic acetylcholine receptors mediate beta-amyloid peptide-induced tau protein phosphorylation. J. Biol. Chem. 2003, 278, 31547–31553.
- Liu, Q.; Xie, X.; Lukas, R.J.; John, P.A.S.; Wu, J. A novel nicotinic mechanism underlies beta-amyloidinduced neuronal hyperexcitation. J. Neurosci. 2013, 33, 7253–7263.
- Dineley, K.T.; Westerman, M.; Bui, D.; Bell, K.; Hsiao Ashe, K.; Sweatt, J.D. β-amyloid activates the mitogen-activated protein kinase cascade via hippocampal a7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J. Neurosci. 2001, 21, 4125–4133.
- Wang, H.Y.; Lee, D.H.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. β-Amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity: Implications for Alzheimer’s disease pathology. J. Biol. Chem. 2000, 275, 5626–5632.
- Ono, K.; Hasegawa, K.; Yamada, M.; Naiki, H. Nicotine breaks down preformed Alzheimer’s β-amyloid fibrils in vitro. Biol. Psychiatry 2002, 52, 880–886.
- Nordberg, A.; Hellström-Lindahl, E.; Lee, M.; Johnson, M.; Mousavi, M.; Hall, R.; Perry, E.; Bednar, I.; Court, J. Chronic nicotine treatment reduces β-amyloidosis in the brain of a mouse model of Alzheimer’s disease (APPsw). J. Neurochem. 2002, 81, 655–658.
- Geula, C.; Mesulam, M. Special properties of cholinesterases in the cerebral cortex of Alzheimer’s disease. Brain Res. 1989, 498, 185–189.
- Sberna, G.; Sáez-Valero, J.; Beyreuther, K.; Masters, C.L.; Small, D.H. The amyloid beta-protein of Alzheimer’s disease increases acetylcholinesterase expression by increasing intracellular calcium in embryonal carcinoma P19 cells. J. Neurochem. 1997, 69, 1177–1184.
- Trang, A.; Khandhar, P.B. Physiology, Acetylcholinesterase. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK539735/ (accessed on 23 May 2021).
- Fodero, L.R.; Mok, S.S.; Losic, D.; Martin, L.L.; Aguilar, M.I.; Barrow, C.J.; Livett, B.G.; Small, D.H. α7-Nicotinic acetylcholine receptors mediate an Aβ1−42-induced increase in the level of acetylcholinesterase in primary cortical neurones. J. Neurochem. 2004, 88, 1186–1193.
- Lazarevic-Pasti, T.; Leskovac, A.; Momic, T.; Petrovic, S.; Vasic, V. Modulators of acetylcholinesterase activity: From Alzheimer’s disease to anti-cancer drugs. Curr. Med. Chem. 2017, 24, 3283–3309.
- Liu, Q.; Kawai, H.; Berg, D.K. Beta-amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 4734–4739.
- Puzzo, D.; Privitera, L.; Leznik, E.; Fà, M.; Staniszewski, A.; Palmeri, A.; Arancio, O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 2008, 28, 14537–14545.
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2011, 11, 155–161.
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172.
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343.
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43.
- Echeverria, V.; Yarkov, A.; Aliev, G. Positive modulators of the α7 nicotinic receptor against neuroinflammation and cognitive impairment in Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 142–157.
- Maelicke, A. Allosteric modulation of nicotinic receptors as a treatment strategy for Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2000, 11, 11–18.
- Weltzin, M.M.; Schulte, M.K. Desformylflustrabromine Modulates α4β2 Neuronal Nicotinic Acetylcholine Receptor High- and Low-Sensitivity Isoforms at Allosteric Clefts Containing the β2 Subunit. J. Pharmacol. Exp. Ther. 2015, 354, 184–194.
- Razay, G.; Wilcock, G.K. Galantamine in Alzheimer’s disease. Expert. Rev. Neurother. 2008, 8, 9–17.
- Echeverria, V.; Zeitlin, R. Cotinine: A potential new therapeutic agent against Alzheimer’s disease. CNS Neurosci. Ther. 2012, 18, 517–523.
- Jiang, S.; Li, Y.; Zhang, C.; Zhao, Y.; Bu, G.; Xu, H.; Zhang, Y.W. M1 muscarinic acetylcholine receptor in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 295–307.
- Abrams, P.; Andersson, K.E.; Buccafusco, J.J.; Chapple, C.; de Groat, W.C.; Fryer, A.D.; Kay, G.; Laties, A.; Nathanson, N.M.; Pasricha, P.J.; et al. Muscarinic receptors: Their distribution and function in body systems, and the implications for treating overactive bladder. Br. J. Pharmacol. 2006, 148, 565–578.
- Felder, C.C.; Bymaster, F.P.; Ward, J.; DeLapp, N. Therapeutic opportunities for muscarinic receptors in the central nervous system. J. Med. Chem. 2000, 43, 4333–4353.
- Conn, P.J.; Jones, C.K.; Lindsley, C.W. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol. Sci. 2009, 30, 148–155.
- Kudlak, M.; Tadi, P. Physiology, Muscarinic Receptor. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Volpicelli, L.A.; Allan, I.L. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog. Brain Res. 2004, 145, 59–66.
- Ishii, M.; Kurachi, Y. Muscarinic acetylcholine receptors. Curr. Pharm. Des. 2006, 12, 3573–3581.
- Miyakawa, T.; Yamada, M.; Duttaroy, A.; Wess, J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J. Neurosci. 2001, 21, 5239–5250.
- Caccamo, A.; Oddo, S.; Billings, L.M.; Green, K.N.; Martinez-Coria, H.; Fisher, A.; LaFerla, F.M. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 2006, 49, 671–682.
- Shinoe, T.; Matsui, M.; Taketo, M.M.; Manabe, T. Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. J. Neurosci. 2005, 25, 11194–11200.
- Fisher, A. Cholinergic treatments with emphasis on m1 muscarinic agonists as potential disease-modifying agents for Alzheimer’s disease. Neurotherapeutics 2008, 5, 433–442.
- Lebois, E.P.; Thorn, C.; Edgerton, J.R.; Popiolek, M.; Xi, S. Muscarinic receptor subtype distribution in the central nervous system and relevance to aging and Alzheimer’s disease. Neuropharmacology 2018, 136, 362–373.
- Caporaso, G.L.; Gandy, S.E.; Buxbaum, J.D.; Ramabhadran, T.V.; Greengard, P. Protein phosphorylation regulates secretion of Alzheimer beta/A4 amyloid precursor protein. Proc. Natl. Acad. Sci. USA 1992, 89, 3055–3059.
- Buxbaum, J.D.; Oishi, M.; Chen, H.I.; Pinkas-Kramarski, R.; Jaffe, E.A.; Gandy, S.E.; Greengard, P. Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc. Natl. Acad. Sci. USA 1992, 89, 10075–10078.
- Davis, A.A.; Fritz, J.J.; Wess, J.; Lah, J.J.; Levey, A.I. Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo. J. Neurosci. 2010, 30, 4190–4196.
- Clader, J.W.; Wang, Y. Muscarinic receptor agonists and antagonists in the treatment of Alzheimer’s disease. Curr. Pharm. Des. 2005, 11, 3353–3361.
- English, B.A.; Webster, A.A. Acetylcholinesterase and its Inhibitors. In Primer on the Autonomic Nervous System; Academic Press: Cambridge, MA, USA, 2012; pp. 631–633.
- Hoskin, J.L.; Al-Hasan, Y.; Sabbagh, M.N. Nicotinic acetylcholine receptor agonists for the treatment of Alzheimer’s dementia: An update. Nicotine Tob. Res. 2019, 21, 370–376.
This entry is offline, you can click here to edit this entry!