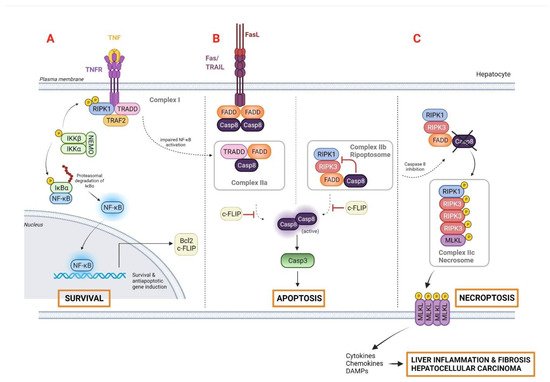
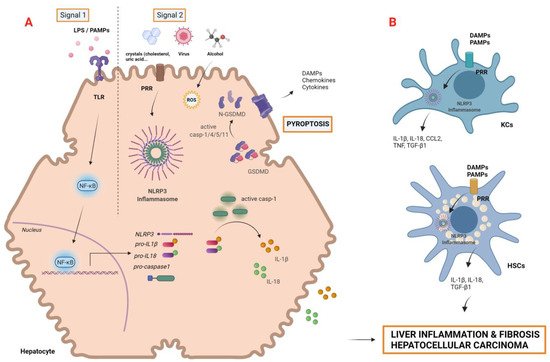
Autophagy is an intracellular lysosomal pathway that plays a pivotal role in homeostasis maintenance in diverse physiological processes. Considered as a “self-eating” process, capable of resisting metabolic stress by recycling cellular components, autophagy has a controversial dual function in the pathophysiology of liver cancer [
65]. The up- and downregulation of this catabolic mechanism was described in HCC, suggesting that autophagy may act as both a tumor promotor and suppressor during malignancy [
66]. Furthermore, studies based on the inhibition of autophagy demonstrated that basal autophagy plays a suppressive role in the initial dysplastic stage by maintaining genomic stability, removing damaged mitochondria (mitophagy) and preventing malignant transformation with accumulated mutations [
67]. However, once the tumor is established, in a proliferative stage, autophagy would promote tumorigenesis, supporting cell growth [
68]. p62, a ubiquitin-binding autophagy receptor, is accumulated in premalignant liver diseases and most HCCs. Its expression is needed and sufficient for activation of the transcription factor NRF2 and mTORC1, the induction of c-Myc, and the protection of HCC-initiating cells from oxidative-stress-induced death [
69]. Importantly, autophagy sustains an oxidative metabolism, which is required for tumor development. In a liver damage scenario, active autophagy promotes reactive oxygen species (ROS) generation, resulting in an upregulated oxidative stress that may induce cell death, followed by compensatory cell proliferation [
70]. Interestingly, the impact of autophagy-mediated metabolic reprogramming on therapeutic resistance is largely unclear, especially in liver cancer [
71,
72,
73,
74]. Sorafenib, an oral multi-kinase inhibitor that is widely used in advanced-stage HCC, results in a significant prolonged survival rate in liver cancer patients [
71]. Nevertheless, a considerable number of HCC patients are refractory to sorafenib treatment due to, at least, the potential induction of autophagy through different mechanisms, such as the activation of the AMPK/mTOR signaling pathway or FOXO-3 mRNA methylation and stabilization [
72,
73,
74]. In that sense, strategies focused on autophagy modulation, combined with sorafenib, have been recently described as new therapeutic options with which to overcome drug resistance in HCC [
75,
76,
77].