Cancer is one of the most extreme medical conditions in both developing and developed countries around the world, causing millions of deaths each year. Chemotherapy and/or radiotherapy are key for treatment approaches, but both have numerous adverse health effects. Furthermore, the resistance of cancerous cells to anticancer medication leads to treatment failure. The rising burden of cancer overall requires novel efficacious treatment modalities. Natural medications offer feasible alternative options against malignancy in contrast to western medication.
This review highlights the potential for furanocoumarins to be clinically beneficial in cancer, particularly given their specificity to tumor cells (while sparing normal cells). In vitro investigations have shown that furanocoumarins affect a range of cellular mechanisms, such as apoptosis, autophagy, and cell cycle arrest. ER stress induction mainly caused by NF-κB inactivation, PI3K/Akt inhibition, and p53 modulation. Furanocoumarins are also effective in different MDR cancers that are the main cause of anticancer therapeutics failure.
Compounds in this class have also have been shown to positively synergize with commonly used anticancer drugs. The fast absorption of furanocoumarins from food into the human bloodstream is also noteworthy. Furanocoumarins, by inhibiting CYP P450 3A4, not only have anticancer properties but also when co-administered with a low bioavailability anticancer compound can increase oral bioavailability. To date, most focus has been on in vitro studies, making it hard to reach solid conclusions on the efficacy of furanocoumarins in vivo.
Nonetheless, studies aimed at characterizing furanocoumarin’s efficacy in vivo as well as clinical studies are encouraging, supporting the need for future studies to better characterize furanocoumarin’s potential as efficacious anticancer treatment modalities.
- furanocoumarin
- apoptosis
- autophagy
- metastasis
- cell cycle arrest
- Anticancer
- Herbal medicine
- Phytopharmacology
- NF-κB Inactivation
- PI3K/Akt Inhibition
- p53 Modulation
- Antioxidant
- MDR Cancers
1. Introduction
Cancer exacts one of the greatest medical tolls on humankind, requiring a proactive procedure for prevention and treatment. An enormous number of patients succumb to cancer every year. It is one of the chief reasons for mortality around the world, and the number of cases is continually expanding and estimated to reach 21 million by 2030. The lack of efficient anticancer treatments remains a clinical problem [1][2]. Chemotherapy and/or radiotherapy are the main clinical approaches to cancer treatment, yet both have documented adverse effects [3][4][5][6]. Cancer treatment affects not only rapidly multiplying cancerous cells but also normal body cells (bone marrow, gastrointestinal tract (GIT), and hair follicles); therefore, these treatments may give rise to severe adverse symptoms. Moreover, quick disposal and widespread distribution of the medications in cancer-free organs requires high dosing, which may lead to incremental adverse reactions. Resistance towards malignant growth is another restriction.
Restorative plants have been utilized previously. Phytopharmaceuticals primarily target malignant growth, and hence, they are the most appropriate contender for anticancer medications [1][2]. Nowadays, significant efforts have improved the efficacy of natural anticancer drugs with the appearance of encouraging strategies [7][8].
Furanocoumarins are phytochemicals that have been utilized for quite a while. The Atharva-Veda, the Indian hallowed book, portrays the Psoralea corylifolia poultice, and the old Egyptians utilized Ammi majus for leukoderma (vitiligo). In 1838, 5-Methoxypsoralen (5-MOP) was the first furanocoumarin isolated from Citrus bergamia oil by Kalbrunner [9]. Furocoumarins are formed by coumarin and a furan ring combination, resulting in angular or linear isomers depending on the furan ring position. Angelicin and psoralen are basic furocoumarins that act as precursors for angular and linear furocoumarins, respectively. These compounds are, for the most part, biosynthesized by phenylpropanoid and the mevalonic pathways. Furocoumarins are produced in plants of Apiaceae and Rutaceae as well as in Asteraceae, Caryophyllaccae, Fabaceae, Moraceae, and Salvadoraceae for defense against insects, bacterial and fungal predators. They provide antimicrobial and insecticidal activity and behave as natural pesticides [10]. Furocoumarins have promising therapeutic prospects, such as analgesic, anticonvulsive, anticoagulant, hypotensive, antidepressants, antibacterial, antifungal, antiviral, anti-inflammatory, antiallergic [11][12], antioxidants [13], and inhibitors of human carbonic anhydrase isozymes [14], against skin diseases [15][16], hyperproliferative disorders [17][18] and as an anticancer [19]. This review is aimed at evaluating the literature on the anticancer potential of various furanocoumarins through different underlying mechanisms and thereof therapeutic/clinical status.
2. Chemistry of Furocoumarins
The exact molecular mechanism of such an activity relies upon the chemical structure of furanocoumarins, which depends on the furan ring and coumarin backbone combination in an angular or linear structure just as the type, location, and the number of the substituents attached [11]. The CH3 presence at C5 improves the tumor properties of psoralen and 5-MOP, paying little heed to the substituent location. The substitution of the methoxy group with an isopentenyloxy moiety in the C5 position prompted abatement in the pro-apoptotic properties of the compound [20,21,22]. Angelicin is the most straightforward angular furanocoumarin and it displays counter cancer properties. Analogous to linear furocoumarins, angular analogs can be substituted with a methoxy or isopentenyloxy group. Methoxy subordinates of angelicin incorporate isobergapten and sphondin. Isobergapten, for example, 5-methoxyangelicine, is a linear isomer of bergapten with a methoxy group joined to the fifth (C5) carbon atom. Thus, sphondin (6-methoxyangelicin) can be considered as an angular analogue of xanthotoxin. The thing that matters is, be that as it may, that the methoxy group is appended to the C6 position in 6-methoxyangelicin and to the C8 atom in the 8-MOP [11].
Furanocoumarins’ defensive and restorative properties have been observed in leukemia, glioma, breast, lung, renal, liver, colon, cervical, ovarian, and prostate malignancies. Apoptosis, autophagy, antioxidant, cell cycle capture, Nuclear Factor Kappa-light-chain-enhancer of activated B cells (NF-κB) inactivation, Phosphatidylinositol 3-kinase/RAC-α Serine/Threonine-Protein Kinase (PI3K/Akt) inhibition, and p53 modulation incorporate mechanistic insight (Figure 1). In this article, we have reviewed the experimental data showed the role of furanocoumarins for cancer prevention and treatment.
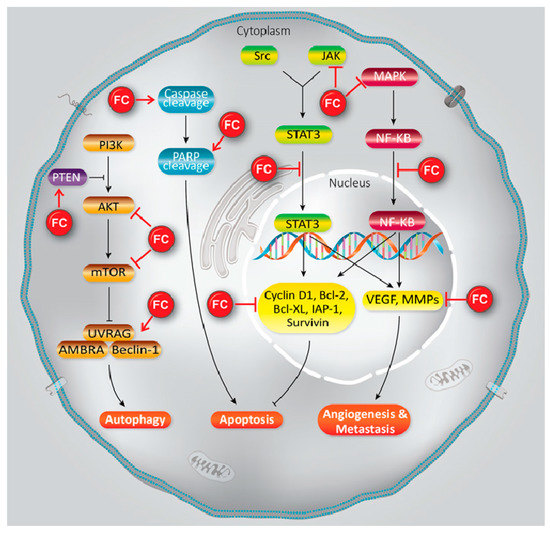
Figure 1. A schema of different molecular mechanisms that are targeted by furanocoumarin. It shows several molecular singling pathways modulation that leads to autophagy, apoptosis, angiogenesis, and metastasis. Black lines: induce, and red lines: inhibit.
3. Bioavailability of Furocoumarins
Furanocoumarins are rapidly absorbed from food into the human bloodstream and detected in plasma within 2–15 min after administration [20], and distributed to the skin, blood, liver, brain, spleen, kidney, and testis. In plasma, furanocoumarins bind to albumins and other plasma proteins. Furanocoumarins are metabolized to psoralen and isopsoralen by intestinal bacteria in the digestive tract. Then, furanocoumarins are excreted into urine as hydroxylated or glucuronated products within 1 h after ingestion. They remain in urine as long as 24 h post-administration. It was also observed that furanocoumarins are converted to bergaptol before excretion [11][21][22].
A significant advance in the investigation of the anticancer properties of furanocoumarins was the revelation of their antiproliferative activity arresting cell-cycle capture and causing cell death [23][24][25]. At the cellular levels, furanocoumarins appear to affect actin filaments, which might be valuable in metastasis prevention [26].
4. Role in MDR Cancers
Cancer cell defiance to chemotherapy is one of the significant deterrents for counter cancer medications due to the involvement of different mechanisms. The main causes are related to the increment of multidrug efflux pumps, including P-gp by tumor cells, and showed MDR. Coumarins have a significant role in MDR inversion [27][28][29].
Psoralen inverts the P-gp-instigated MDR in MCF-7/ADR by repressing the efflux capacity of P gp [29] and by hindrance of P-gp ATPase activity. Similarly, isopimpinellin (IC50 value 26 µM) and Phellopterin (IC50 value 8 µM) exhibited cytotoxic activity against MDR HL-60/MX2 (human promyelocytic leukemia cells) and CEM/C1 (human lymphoblastic leukemia cells) respectively [30]. Feroniellin A reduces MDR in A549 cell lines by decreasing P-gp. MDR1rapidly pumps out anticancer drugs, decreases intracellular drug concentrations and leads to the failure of anticancer therapeutics [31]. Similarly, BCRP and MRP overexpression participates in MDR [32][33]. Bergapten and methoxsalen showed cytotoxicity in MDR1, MRP2 BCRP overexpressing gastric (EPG85.257RDB), ovarian (A2780RCIS) and breast (MCF7MX) cancer cell lines which showed reticence of MDR1, BCRP, and MRP efflux functions [34].
ETRα positive and estrogen dependence are seen in 70% of breast cancers. ETRα depletion during initial stages of breast cancer is a potent anticancer approach [35]. Xanthotoxol, bergapten, angelicin, psoralen and isoimperatorin antagonized ETRα activity in MCF-7 cells with IC50 values of 0.72, 1.18, 11.02, 24.08 and 54.32 μM, respectively [36].
Exosomes, secreted from tumor cells to promote tumor progression, such as metastasis and MDR, activate sequestration of anticancer drugs by MDR-1 and P-gp [37]. Psoralen significantly reduces the number of exosomes, which correlates with increased MCF-7/ADR cells’ sensitivity for apoptosis under the influence of chemotherapy. Similar observations were seen in A 549/D16 lung cancer cell lines [38].
5. Furanocoumarins as Adjuvant with Other Anticancer Agents
Invasion and angiogenesis are important targets of FC (Figure 2). In a study, given that bergamottin (50 μM)-potentiated simvastatin (SV) inhibits TNF-induced NF-κB activation and IκBα deterioration, cell-cycle capture in S-phase by boost of p21 and p27 in KBM-5 cells. This impact of co-treatment is associated with decrease incyclin D1 (cell proliferation), Bcl-2, cIAP-1, Bcl-xL and survivin (cell survival), MMP-9 (invasion), and VEGF (angiogenesis) which are directed by NF-κB. CYP P450 34A inhibition by bergamottin enhances SV bioavailability in KBM-5 to synergize SV effects [39].Cytochrome P450 upregulation in cancer makes it one of the effective strategy against it. CYP P450 inhibition by furanocoumarins and other phytoconstituents leads to anticarcinogenic effects, down regulating MDR and prolonging the t1/2 of other anticancer drugs [40][41]. Bergamottin, DHBG, bergapten, and bergaptol inhibit CYP P450 34A to potentiate vinblastine effects [42]. Bergamottin, imperatorin, and isopimpinellin repressed CYP P450-1A1, -1A2, -3A4, and -1B1 (involve in carcinogen metabolism), in the MCF-7 cell lines [43]. MDR has been linked to BCRP, MRP, and MDR1 increase that expel cisplatin, daunorubicin, and mitoxantrone are very common in cancer therapeutics [35]. Bergapten and methoxsalen are capable of averting mitoxantrone, cisplatin, and daunorubicin binding to MDR1, BCRP, and MRP and, therefore, impeding their cellular efflux [35]. Bergamottin, DHBG, bergapten, and bergaptol from grapefruit juice inhibit P-gp and MRP2 mediated vinblastine efflux [41]. Long-term use of tamoxifen in breast cancer leads to MDR and an increased risk of endometrial carcinogenesis [44]. Bergapten degrades and depletes ETRα from MCF-7 tamoxifen-resistant cells by inducing SMAD4 protein to block mitogenic signals . The anticancer effects of doxorubicin in HeLa cells are enhanced by Imperatorin via Mcl-1 down-regulation [45]. Imperatorin (100 μM) effectively increasing autophagy in HeLa cells in response to cisplatin (2 μM) by microtubule-associated protein 1A/1B-light chain 3 (LC3) cleavage and the inhibition of Hsp27 and Hsp72[46] . Imperatorin potentiates the cytotoxicity of cisplatin in hepatocellular carcinoma (HCC) cells (HepG2, HepG3B, PLC, Huh7) by downregulating Mcl-1 expression [47].
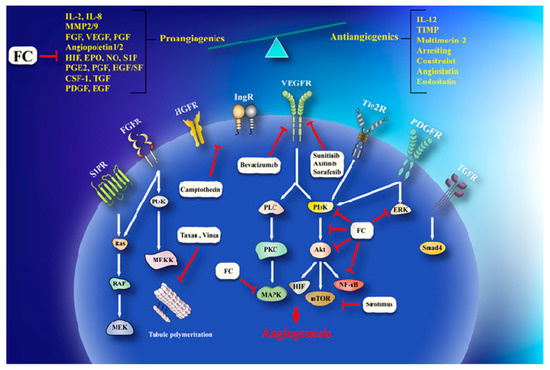
Figure 2. A schema of anti-angiogenesis effects of furanocoumarin. FC via activation/inhibition of a sequences of cellular and molecular pathways exerts its anti-angiogenesis effects. Black lines: induce, and red lines: inhibit.
6. Furanocoumarins and Cancer Risks
PUVA therapy (photoactivated psoralens) is beneficial for patients with vitiligo, psoriasis, and skin diseases. PUVA activates caspase-3, -8 and -9 in Jurkat (human tumor T-cell line) [48]. PUVA, the photoactivated psoralens (5-MOP and 8-MOP) induce apoptosis in MCF-7, ZR-75 and SKBR-3 by caspase and p53 upregulation, PI3K/AKT downregulation and effective against breast hormone-responsive cancers [49]. PUVA arrest G2/M phase by phospho-Chk1 increment and phospho-Cdc2 decrement in B16F10 murine melanoma cell line [50]. Unfortunately, it is linked toa risk of basal cell, squamous cell, and non-melanoma skin cancers [51][52]. PUVA has been approved by the FD, [53]. PUVA is ideal for superficial applications only, due to low penetration into deep tissues. Therefore, X-PACT has been introduced, which at low X-ray doses photoactivates psoralens and hence mitigates the low penetration problem and adverse effects of PUVA [54].
7. Conclusions
Chemotherapy and radiation are the staples for treatment of malignant growth, however, both have serious adverse health symptoms. It is known that tumors have developed many mechanisms at the molecular level enabling cell survival during chemotherapy. Therefore, it is essential to develop novel pharmaceuticals with increased efficacy and reduced toxicity. This review highlights the potential for furanocoumarins to be clinically beneficial in cancer, particularly given their specificity to tumor cells (while sparing normal cells). In vitro investigations have shown that furanocoumarins affect a range of cellular mechanisms, such as apoptosis, autophagy, and cell cycle arrest. ER stress induction mainly caused by NF-κB inactivation, PI3K/Akt inhibition, and p53 modulation. Furanocoumarins are also effective in different MDR cancers that are the main cause of anticancer therapeutics failure. Compounds in this class have also have been shown to positively synergize with commonly used anticancer drugs. The fast absorption of furanocoumarins from food into the human bloodstream is also noteworthy [55]. Furanocoumarins, by inhibiting CYP P450 3A4, not only have anticancer properties but also when co-administered with a low bioavailability anticancer compound can increase oral bioavailability [56]. Thus, as to improve genuine treatments for various sorts of tumors, nanomedicine has developed new strategies coordinated to build the efficacy of medications focusing on tumors and limit their side effects [57][58]. Furanocoumarin-loaded lipid-polymer-hybrid-nanoparticles represent an additional option for sustained release of these molecules to improve efficacy and synergistic effects with other anticancer agents and against MDR cancers [30][59][60]. To date, most focus has been on in vitro studies, making it hard to reach solid conclusions on the efficacy of furanocoumarins in vivo. Nonetheless, studies aimed at characterizing furanocoumarin’s efficacy in vivo as well as clinical studies are encouraging, supporting the need for future studies to better characterize furanocoumarin’s potential as efficacious anticancer treatment modalities.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21165622
References
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield Trends Are Insufficient to Double Global Crop Production by 2050. PLoS ONE 2013, 108, 20260-4.
- Röös, E.; Bajželj, B.; Smith, P.; Patel, M.; Little, D.; Garnett, T. Greedy or needy? Land use and climate impacts of food in 2050 under different livestock futures. Glob. Environ. Chang. 2017, 47, 1–2.
- Zhang, H.; Zhang, J.; Lang, Z.; Botella, J.R.; Zhu, J.K. Genome Editing—Principles and Applications for Functional Genomics Research and Crop Improvement. CRC. Crit. Rev. Plant Sci. 2017, 36, 291–309.
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405.
- Rai, K.M.; Ghose, K.; Rai, A.; Singh, H.; Srivastava, R.; Mendu, V. Genome engineering tools in plant synthetic biology. In Current Developments in Biotechnology and Bioengineering: Synthetic Biology. Cell Eng. Bioprocess. Technol. 2018, doi:10.1016/B978-0-444-64085-7.00003-4
- Osakabe, K.; Osakabe, Y.; Toki, S. Site-directed mutagenesis in Arabidopsis using custom-designed zinc finger nucleases. Proc. Natl. Acad. Sci. USA 2010, 107, 12034–12039.
- Petolino, J.F.; Worden, A.; Curlee, K.; Connell, J.; Moynahan, T.L.S.; Larsen, C.; Russell, S. Zinc finger nuclease-mediated transgene deletion. Plant Mol. Biol. 2010, 73, 617–628.
- Shukla, V.K.; Doyon, Y.; Miller, J.C.; Dekelver, R.C.; Moehle, E.A.; Worden, S.E.; Mitchell, J.C.; Arnold, N.L.; Gopalan, S.; Meng, X.; et al. Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature 2009, 459, 437–441.
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512.
- Li, T.; Liu, B.; Spalding, M.H.; Weeks, D.P.; Yang, B. High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 2012, 30, 390.
- Kopischke, S.; Schüßler, E.; Althoff, F.; Zachgo, S. TALEN-mediated genome-editing approaches in the liverwort Marchantia polymorpha yield high efficiencies for targeted mutagenesis. Plant Methods 2017, 13, 20.
- Bortesi, L.; Fischer, R. The CRISPR/Cas9 system for plant genome editing and beyond. Biotechnol. Adv. 2015, 33, 41–52.
- Zhang, Y.; Pribil, M.; Palmgren, M.; Gao, C. A CRISPR way for accelerating improvement of food crops. Nat. Food 2020, 1, 200–205, doi:10.1038/s43016-020-0051-8.
- Butt, H.; Zaidi, S.S. e. A.; Hassan, N.; Mahfouz, M. CRISPR-Based Directed Evolution for Crop Improvement. Trends Biotechnol. 2020, 38, 236–240, doi:10.1016/j.tibtech.2019.08.001.
- Puchta, H. Applying CRISPR/Cas for genome engineering in plants: The best is yet to come. Curr. Opin. Plant Biol. 2017, 36, 1–8.
- Ahmar, S.; Gill, R.A.; Jung, K.H.; Faheem, A.; Qasim, M.U.; Mubeen, M.; Zhou, W. Conventional and molecular techniques from simple breeding to speed breeding in crop plants: Recent advances and future outlook. Int. J. Mol. Sci. 2020, 21, 2590.
- Mishra, R.; Zhao, K. Genome editing technologies and their applications in crop improvement. Plant Biotechnol. Rep. 2018, 12, 57–68.
- Townsend, J.A.; Wright, D.A.; Winfrey, R.J.; Fu, F.; Maeder, M.L.; Joung, J.K.; Voytas, D.F. High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature 2009, 459, 442–445.
- Zhang, F.; Maeder, M.L.; Unger-Wallaced, E.; Hoshaw, J.P.; Reyon, D.; Christian, M.; Li, X.; Pierick, C.J.; Dobbs, D.; Peterson, T.; et al. High frequency targeted mutagenesis in Arabidopsis thaliana using zinc finger nucleases. Proc. Natl. Acad. Sci. USA 2010, 107, 12028–12033.
- Puchta, H. The repair of double-strand breaks in plants: Mechanisms and consequences for genome evolution. J. Exp. Bot. 2005, 56, 1–4.
- Durai, S.; Mani, M.; Kandavelou, K.; Wu, J.; Porteus, M.H.; Chandrasegaran, S. Zinc finger nucleases: Custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res. 2005, 33, 5978–5990.
- Papworth, M.; Kolasinska, P.; Minczuk, M. Designer zinc-finger proteins and their applications. Gene 2006, 366, 27–38.
- Carlson, D.F.; Fahrenkrug, S.C.; Hackett, P.B. Targeting DNA with fingers and TALENs. Mol. Ther. Nucleic Acids 2012, doi:10.1038/mtna.2011.5.
- Carroll, D.; Morton, J.J.; Beumer, K.J.; Segal, D.J. Design, construction and in vitro testing of zinc finger nucleases. Nat. Protoc. 2006, 1, 1329–1341.
- Minczuk, M.; Papworth, M.A.; Miller, J.C.; Murphy, M.P.; Klug, A. Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res. 2008, 36, 3926–3938.
- Gaj, T.; Guo, J.; Kato, Y.; Sirk, S.J.; Barbas, C.F. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat. Methods 2012, 9, 805–807.
- Zhou, H.; Liu, B.; Weeks, D.P.; Spalding, M.H.; Yang, B. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice. Nucleic Acids Res. 2014, 42, 10903–10914.
- Xu, R.F.; Li, H.; Qin, R.Y.; Li, J.; Qiu, C.H.; Yang, Y.C.; Ma, H.; Li, L.; Wei, P.C.; Yang, J.B. Generation of inheritable and “transgene clean” targeted genome-modified rice in later generations using the CRISPR/Cas9 system. Sci. Rep. 2015, 19, 11491.
- Lawrenson, T.; Shorinola, O.; Stacey, N.; Li, C.; Østergaard, L.; Patron, N.; Uauy, C.; Harwood, W. Induction of targeted, heritable mutations in barley and Brassica oleracea using RNA-guided Cas9 nuclease. Genome Biol. 2015, 16, 258.
- Morineau, C.; Bellec, Y.; Tellier, F.; Gissot, L.; Kelemen, Z.; Nogué, F.; Faure, J.D. Selective gene dosage by CRISPR-Cas9 genome editing in hexaploid Camelina sativa. Plant Biotechnol. J. 2017, 15, 729–739.
- Okuzaki, A.; Ogawa, T.; Koizuka, C.; Kaneko, K.; Inaba, M.; Imamura, J.; Koizuka, N. CRISPR/Cas9-mediated genome editing of the fatty acid desaturase 2 gene in Brassica napus. Plant Physiol. Biochem. 2018, 131, 63–69.
- Long, S.P.; Marshall-Colon, A.; Zhu, X.G. Meeting the global food demand of the future by engineering crop photosynthesis and yield potential. Cell 2015, 161, 56–66.
- Shen, L.; Wang, C.; Fu, Y.; Wang, J.; Liu, Q.; Zhang, X.; Yan, C.; Qian, Q.; Wang, K. QTL editing confers opposing yield performance in different rice varieties. J. Integr. Plant Biol. 2018, 60, 89–93.
- Li, R.; Li, R.; Li, X.; Fu, D.; Zhu, B.; Tian, H.; Luo, Y.; Zhu, H. Multiplexed CRISPR/Cas9-mediated metabolic engineering of γ-aminobutyric acid levels in Solanum lycopersicum. Plant Biotechnol. J. 2018, 16, 415–427.
- Bertier, L.D.; Ron, M.; Huo, H.; Bradford, K.J.; Britt, A.B.; Michelmore, R.W. High-resolution analysis of the efficiency, heritability, and editing outcomes of CRISPR/Cas9-induced modifications of NCED4 in lettuce (Lactuca sativa). G3 Genes Genomes Genet. 2018, 8, 1513–1521.
- Ueta, R.; Abe, C.; Watanabe, T.; Sugano, S.S.; Ishihara, R.; Ezura, H.; Osakabe, Y.; Osakabe, K. Rapid breeding of parthenocarpic tomato plants using CRISPR/Cas9. Sci. Rep. 2017, 7, 507.
- Soyk, S.; Müller, N.A.; Park, S.J.; Schmalenbach, I.; Jiang, K.; Hayama, R.; Zhang, L.; Van Eck, J.; Jiménez-Gómez, J.M.; Lippman, Z.B. Variation in the flowering gene SELF PRUNING 5G promotes day-neutrality and early yield in tomato. Nat. Genet. 2017, 49, 162–168.
- Brooks, C.; Nekrasov, V.; Lippman, Z.B.; Van Eck, J. Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 system. Plant Physiol. 2014, 166, 1292–1297.
- Zhou, J.; Li, D.; Wang, G.; Wang, F.; Kunjal, M.; Joldersma, D.; Liu, Z. Application and future perspective of CRISPR/Cas9 genome editing in fruit crops. J. Integr. Plant Biol. 2020, 62, 269–286.
- Jia, H.; Nian, W. Targeted genome editing of sweet orange using Cas9/sgRNA. PLoS ONE 2014, 9, e93806.
- Malnoy, M.; Viola, R.; Jung, M.H.; Koo, O.J.; Kim, S.; Kim, J.S.; Velasco, R.; Kanchiswamy, C.N. DNA-free genetically edited grapevine and apple protoplast using CRISPR/Cas9 ribonucleoproteins. Front. Plant Sci. 2016, 7, 1904.
- Toda, E.; Koiso, N.; Takebayashi, A.; Ichikawa, M.; Kiba, T.; Osakabe, K.; Osakabe, Y.; Sakakibara, H.; Kato, N.; Okamoto, T. An efficient DNA- and selectable-marker-free genome-editing system using zygotes in rice. Nat. Plants 2019, 5, 363.
- Espley, R.V.; Leif, D.; Plunkett, B.; McGhie, T.; Henry-Kirk, R.; Hall, M.; Johnston, J.W.; Punter, M.P.; Boldingh, H.; Nardozza, S.; et al. Red to Brown: An Elevated Anthocyanic Response in Apple Drives Ethylene to Advance Maturity and Fruit Flesh Browning. Front. Plant Sci. 2019, 10, 1248.
- Klap, C.; Yeshayahou, E.; Bolger, A.M.; Arazi, T.; Gupta, S.K.; Shabtai, S.; Usadel, B.; Salts, Y.; Barg, R. Tomato facultative parthenocarpy results from SlAGAMOUS-LIKE 6 loss of function. Plant Biotechnol. J. 2017, 15, 634–647.
- Begemann, M.B.; Gray, B.N.; January, E.; Gordon, G.C.; He, Y.; Liu, H.; Wu, X.; Brutnell, T.P.; Mockler, T.C.; Oufattole, M. Precise insertion and guided editing of higher plant genomes using Cpf1 CRISPR nucleases. Sci. Rep. 2017, 7, 11606.
- Wang, M.; Mao, Y.; Lu, Y.; Wang, Z.; Tao, X.; Zhu, J.K. Multiplex gene editing in rice with simplified CRISPR-Cpf1 and CRISPR-Cas9 systems. J. Integr. Plant Biol. 2018, 60, 626–631.
- Naim, F.; Dugdale, B.; Kleidon, J.; Brinin, A.; Shand, K.; Waterhouse, P.; Dale, J. Gene editing the phytoene desaturase alleles of Cavendish banana using CRISPR/Cas9. Transgenic Res. 2018, 27, 451–460.
- Kim, H.; Kim, S.T.; Ryu, J.; Kang, B.C.; Kim, J.S.; Kim, S.G. CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat. Commun. 2017, 8, 14406.
- Xu, R.; Qin, R.; Li, H.; Li, D.; Li, L.; Wei, P.; Yang, J. Generation of targeted mutant rice using a CRISPR-Cpf1 system. Plant Biotechnol. J. 2017, 15, 713–717.
- Jiang, W.; Zhou, H.; Bi, H.; Fromm, M.; Yang, B.; Weeks, D.P. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 2013, 41, e188.
- Li, M.; Li, X.; Zhou, Z.; Wu, P.; Fang, M.; Pan, X.; Lin, Q.; Luo, W.; Wu, G.; Li, H. Reassessment of the four yield-related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Front. Plant Sci. 2016, 7, 377.
- Wang, J.; Cappa, J.J.; Harris, J.P.; Edger, P.P.; Zhou, W.; Pires, J.C.; Adair, M.; Unruh, S.A.; Simmons, M.P.; Schiavon, M.; et al. Transcriptome-wide comparison of selenium hyperaccumulator and nonaccumulator Stanleya species provides new insight into key processes mediating the hyperaccumulation syndrome. Plant Biotechnol. J. 2018, 16, 1582–1594.
- Cai, Y.; Chen, L.; Liu, X.; Guo, C.; Sun, S.; Wu, C.; Jiang, B.; Han, T.; Hou, W. CRISPR/Cas9-mediated targeted mutagenesis of GmFT2a delays flowering time in soya bean. Plant Biotechnol. J. 2018, 16, 176–185.
- Ito, Y.; Nishizawa-Yokoi, A.; Endo, M.; Mikami, M.; Toki, S. CRISPR/Cas9-mediated mutagenesis of the RIN locus that regulates tomato fruit ripening. Biochem. Biophys. Res. Commun. 2015, 467, 76–82
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355.
- Zhang, J.; Zhang, H.; Botella, J.R.; Zhu, J.K. Generation of new glutinous rice by CRISPR/Cas9-targeted mutagenesis of the Waxy gene in elite rice varieties. J. Integr. Plant Biol. 2018, 60, 369–375.
- Li, X.; Zhou, W.; Ren, Y.; Tian, X.; Lv, T.; Wang, Z.; Fang, J.; Chu, C.; Yang, J.; Bu, Q. High-efficiency breeding of early-maturing rice cultivars via CRISPR/Cas9-mediated genome editing. J. Genet. Genomics 2017, 44, 175.
- Qi, W.; Zhu, T.; Tian, Z.; Li, C.; Zhang, W.; Song, R. High-efficiency CRISPR/Cas9 multiplex gene editing using the glycine tRNA-processing system-based strategy in maize. BMC Biotechnol. 2016, 16, 58.
- Andersson, M.; Turesson, H.; Nicolia, A.; Fält, A.S.; Samuelsson, M.; Hofvander, P. Efficient targeted multiallelic mutagenesis in tetraploid potato (Solanum tuberosum) by transient CRISPR-Cas9 expression in protoplasts. Plant Cell Rep. 2017, 36, 117–128.
- Li, A.; Jia, S.; Yobi, A.; Ge, Z.; Sato, S.J.; Zhang, C.; Angelovici, R.; Clemente, T.E.; Holding, D.R. Editing of an alpha-kafirin gene family increases digestibility and protein quality in sorghum. Plant Physiol. 2018, 177, 1425–1438.