4. Discussion
The mechanics and physics behind metastasis are frequently underestimated. Studies have been limited to the point of view of the behavior and movement of the cytoskeleton; however, the mechanical cues derived from cell motility can have much more of an impact on tumor progression than anticipated. A better comprehensive approach could highlight the different roles of migration and nuclear squeezing, from their effect on cellular mechanoresponsivity and in epigenetic switch, to their role in genome diversity generation, and finally their implication in inducing the cGAS/STING pathway.
During metastasis, tumor cells need to overcome several barriers in order to access circulation and migrate to new locations. Cancer cells from epithelial origin do not always have intrinsic invasive and migration properties. Thus, the reprogramming of cancer cells is often necessary for a successful metastatic process. The transient embryogenic reprogramming EMT has been associated with the metastatic process, conferring invading and migrating properties [
82], notably by inducing changes in cytoskeleton organization. EMT is a highly plastic process [
83] that often presents a continuum of hybrid epithelial–mesenchymal states. EMT can be modulated by several factors such as the gradient of several cytokines as TGFβ, a well-known EMT inducer, present under inflammation and in the tumor microenvironment [
84]. Another aspect of EMT plasticity can be the mechanical forces. As mentioned before, cells under constriction can sense the constricted area, and undergo nuclear deformation that triggers chromatin loosing [
43], and increases the cytoskeleton forces [
44,
45] to allow a faster escape from the constriction. Those rapid changes in the cytoskeleton can be interpreted as a hybrid E-M state. The mechanosensors from the plasmatic membrane can also sense the matrix stiffness and can modulate the cytoskeleton, as well as the EMT process [
37,
40]. Altogether, such mechanoresponses favor the diversity in EMT hybrid states due to its rapid changes. The diversity of traction movements due to diverse ECM environments such as pore and channels size and presence of fibers can explain the heterogeneity in EMT states observed in cancer patients [
85].
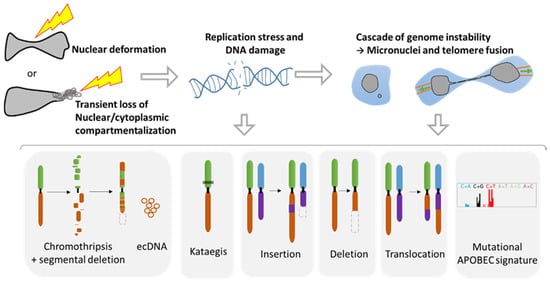
Nuclear squeezing [
46,
47] and extensive forces during migration [
34] can lead to transient NER. Noteworthy, nuclear envelope fragility and NER are also associated to other events [
27] such as viral infection [
86,
87,
88,
89], some neuropathies [
90], aging [
91] or as a consequence of impairment in lamin levels [
91,
92,
93] or function as in some type of laminopathy [
94]. Over the past 10 years, the research on NE biology has revealed unexpected consequences of its deregulation and has shown the implication of NER in unexpected diseases such as in cancer. From a genomic point of view, NER can lead to an extensive genomic diversity, an ally of tumor progression. Resistance against anticancer therapy is acquired through the development of new mutations, chromosomal reorganization and new copy number variations to find new way to bypass the therapy (
Figure 4). Studies from NER of micronuclei and cells with chromatin bridge have shown that NER originates a tremendous variety of genomic reorganizations that are also found during cancer progression (
Figure 3). Each cell can gain new genomic aberrances, increasing the pool of cells with unique genomic combination. It is a lottery and hazardous process that might favor or disfavor growth.
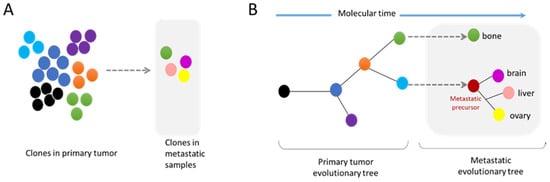
Results from clinic support this premise. Technological advances have allowed the isolation and study of CTC from patients, contributing to the detection of subpopulations that comprise intra-tumor heterogeneity. It also permitted the detection of acquired mutations from metastatic sites that could be used as a diagnostic tool toward personalized medicine during the progression of the disease [
95,
96]. Genomic analysis have shown important heterogeneity between CTC population isolated from the same patient in accordance with the genomic evolution of tumors [
96,
97,
98,
99,
100,
101]. It is likely that the NER that takes place during tumor cell migration is directly involved in the genomic modifications observed in CTC. As human metastases isolated from different sites are enriched in chromosomal instability [
65,
66], it is tempting to speculate that NER plays a major role in the establishment of genomic diversity during tumor progression.
The field of oncology is taking an increasing interest in understanding the tight connection between immune system and cancer cells. Recently two major innate immune mechanisms have come into the limelight. First APOBEC, a DNA mutator participating in the inhibition of retrovirus and retrotransposon mobility. This enzyme binds preferably to DNA stem-loops [
102] and is responsible for one of the most prominent mutation signatures in cancer, present in over half of human tumors, called the APOBEC signature, or Signatures 2/13 and might be also involved in the Aging signature [
103]. APOBEC seems also to be responsible of the Kataegis and appears to be linked with nuclear envelope disruption rupture [
60,
61]. Interestingly, DNA damage and replication stress also seems to stimulate the expression of this enzyme [
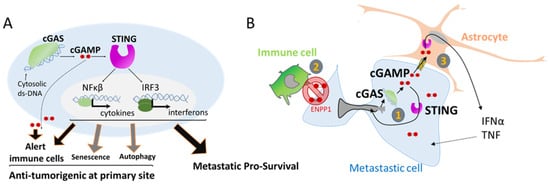
104]. The second, is the cGAS/STING pathway, a recently discovered pathway [
105]. Remarkably, the cGAS/STING pathway, initially involved in tumor cell clearance has also been shown to act as a pro-survival pathway in metastases [
65,
78,
79]. This contradictory action might be a consequence of doses and gradients. In primary tumor sites, levels of extracellular cGAMP and secreted interferon type 1 should be higher than in a metastatic site which only contained few seeded tumor cells. Furthermore, a moderate and transient activation of the innate immune system, as observed during transient NER or due to the limited amount of DNA from micronuclei could be the Achilles heel, by having a protective effect rather than a detrimental one.
Activation of the cGAS/STING pathway leads to the release of several interleukins and interferons in an autocrine or a paracrine fashion. Such signals are indispensable for metastasis survival. Disseminating cells are known to ’talk ‘to the microenvironment and can educate cells around them such as fibroblast (e.g., CAF) to deliver the proper growth factors tailored to their needs. For example, in case of prostate disseminate cells, single cell RNAseq analysis of dormant and proliferative metastatic cells have shown a cross-talk between cancer cells and fibroblasts. Proliferative metastatic cells secrete prostaglandin PGE2 that activates prolactin secretion in nearby fibroblasts, which facilitates tumor cell proliferation [
106]. In the case of the cGAS/STING pathway, cGAMP is a signal that induces the secretion of several pro-inflammatory cytokines such as the senescence-associated secretory phenotype (SASP), known to support angiogenesis and tumorigenesis but as well as interferons type 1 that alert the immune system. Such paradox is still under study and highlight the complexity of the crosstalk between cancer cells and their microenvironment. The growing interest in the cGAS/STING pathway is such that several molecules and strategies to target this route are already delineated and evaluated in preclinical models and in clinical trials for autoimmune diseases [
107,
108]. Because the primary role of cGAS/STING involves tumor clearance, the efforts were focused creating cGAS/STING agonists. However, early results of clinical trials showed limited efficiency [
77], leading to a gear switch to antagonist molecules that could give a better result in blocking metastasis survival [
77,
107]. The surprising link between innate immune system and cancer will reveal new understanding and could lead to new therapeutic targets in the future.
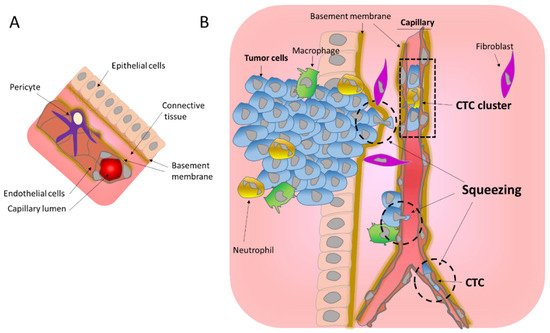
Metastasis is extremely inefficient; it is estimated that less than 1% of cells that intravasate into the blood stream will ultimately succeed in the generation of distant metastasis [
7]. This inefficiency maybe be the result of the mechanical stress excess. The extreme nuclear squeezing during ECM migration, insertion into the transendothelial wall or passage through tiny capillaries can lead to massive DNA damage, that could be deadly or conferring unfavorable mutations. Moreover, metastatic cells can also fail to escape the immune system after cGAS activation. However, mechanical forces may be indispensable for a successful metastasis process. Surprisingly, in a study that fluctuates the density of the BM, it was showed that a BM with loosen connections and bigger pores, impaired the success of metastasis [
1]. This data was also observed in breast cancer patients in which loosen BM is a good prognostic factor [
1]. The mechanism behind this surprising result could be a balance of mechanoresponsivity that prime migration, induction of genomic instability and activation of the cGAS pathway that favors metastasis survival.