Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
As the non-naturally occurring biomolecules, modified DNA/RNA nucleoside and oligonucleotide analogues composed of L-(deoxy)riboses, have been designed and applied as innovative therapeutics with superior plasma stability, weakened cytotoxicity, and inexistent immunogenicity.
- Nucleoside analogues
- Antitumor Agents
- antiviral agents
1. Introduction
Nucleoside analogues have been playing a vital role as a critical chemotherapy of viral infectious diseases [19,20]. Due to the conformational similarity with naturally occurring nucleotides/nucleosides, various carbocyclic nucleoside analogues have been designed, which can specifically recognize the target polymerases [21] or hydrolases [22] and effectively block their biological activities. Particularly, the outbreaks of severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) in 2019 have especially raised the concerns about the deficiency of effective therapeutics and highlighted the importance of developing multiple antiviral strategies due to the fast drug-resistant mutations. Therefore, tremendous efforts have been explored to develop the broad-spectrum nucleoside analogues targeting the SARS-CoV-2-RNA-dependent RNA polymerase [23]. However, the application of wild-type carbocyclic nucleoside analogues is frequently restricted by the arisen cytotoxicity, after the 5′-phosphorylation happens to the nucleoside analogue and this metabolite interferes with various normal cellular enzymes [24]. To seek a novel platform to diminish the unwanted toxicity, the enantiomeric nucleoside analogues containing the unnatural L-configuration have been pioneered.
2. L-Type Neplanocin Compounds as Anti-Norovirus Therapies
9-(trans-2′,trans-3′-dihydroxycyclopent-4′-enyl)-3-deazaadenine (DHCDA) is a neplanocin A analog and functions as an inhibitor of S-adenosylhomocysteine (AdoHcy) hydrolase, with broad-spectrum antiviral potential against vesicular stomatitis virus, vaccinia virus, parainfluenza virus, reovirus, and rotavirus [25]. Inspired by the observations that both D-like and L-like neplanocin derivatives possess potent antiviral activities, Chen lab has pioneered to synthesize a series of DHCDA analogues, all of which contain L-type configuration and an adenine nucleobase but lack the 5′-hydroxyl group for nucleoside kinases to recognize [26].
The synthesized L-neplanocin derivatives contained a cyclopentenol pseudo-sugar and an adenine nucleobase analogue, which were coupled via a Mitsunobu coupling reaction to provide the target compounds (Figure 1). The syntheses of halogenated derivatives, including bromonation at its nucleobase and 5′-fluorination/bromonation at sugar, were also accomplished. These L-like analogues of natural carbocyclic nucleoside neplanocin A were evaluated as potential inhibitors against norovirus and Ebola viruses. Compounds 1a and 2b showed potent antiviral activity against norovirus (EC50 = 4.2 µM and EC50 = 3.0 µM, respectively). Compound 1b was found to possess effective antiviral activity against the Ebola virus (EC50 = 8.3 µg/mL), while the analogues 2a and 2b were completely inactive. With the additional exocyclic derivatizations at the 5′-position, compound 5 displayed the potent activity against Pichinde (EC50 = 0.9 µg/mL) and Tacaribe (EC50 = 1.3 µg/mL). Both viruses are negative single-stranded RNA viruses belonging to arenaviridae, the same family as the Lassa fever virus. Compound 4a and compound 4b were also active against the Ebola virus. Although 4b showed no activity against those two arenaviruses, it is two-fold more active against the Ebola virus than compound 5.
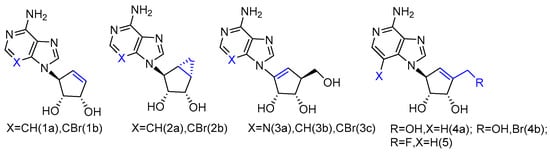
Figure 1. L-like analogues of carbocyclic nucleoside neplanocin.
It is noteworthy that the conformational behavior of “sugar” puckering (north/south) and nucleobase orientation (syn/anti) may contribute to the antiviral activity differences. The crystallographic studies revealed that the sugar in compound 1b adopted a 3′-exo conformation, while the more conformational, rigid bicyclic sugar in compound 2b would lock it into 2′-exo conformation. In addition, because of the steric hindrance, the 3-bromo substitution played an important role in anti-Ebola activity by forcing the nucleobase to adopt the less congested anti-conformation over the syn-conformation. No activity was found for single-stranded positive viruses, which suggests that L-like neplanocin analogues are more effective on single-stranded negative-sense RNA viruses [27].
3. L-Enantiomer of Immucillin Analogue as an Anti-T-Cell Leukaemia Agent
Immucillin-H (ImmH, also known as Forodesine) is a transition-state analog inhibitor of purine nucleoside phosphorylase (PNPases). It has been extensively studied for the treatment of patients with T-cell acute lymphoblastic leukemia (T-ALL), and its C-nucleoside hydrochloride form is in phase II clinical trials as an anti-T-cell leukemia agent [28]. Various Immucillin analogs modified at the 2′-, 3′-, or 5′-positions of the aza sugar moiety or at the 6-, 7-, or 8-positions of the deazapurine, have been synthesized and tested for their inhibition of human PNPases [29]. Inspired by the nucleoside-like structures of Immucillin analogues and their binding modes with PNPases by crystal structures [30,31], their L-enantiomers have been investigated as novel pharmaceuticals against T-cell mediated disorders [32]. The synthetically achieved (1R)-1-(9-Deazahypoxanthin-9-yl)-1,4-dideoxy-1,4-imino-L-ribitol (Figure 2, 6a) was an L-enantiomer of natural D-ImmH, and its hydrochloride complex was revealed to be a slow-onset tight-binding inhibitor of PNPases of human, bovine, and Plasmodium falciparum. Although this compound showed less activity than D-ImmH when inhibiting the selected enzymes, it still demonstrated more excellent binding potency compared to 3′- and 5′-modified D-ImmH. Moreover, the L-enantiomer of second-generation Immucillin analogue, 4′-deaza-1′-aza-2′-deoxy-1′-(9-methylene)-Immucillin-H (DADMe-ImmH) [33], was also synthesized. The L-DADMe-ImmH (Figure 2, 6b) also displayed lower activities as an inhibitor, when binding to the three enzymes. However, it was interesting to observe the sub-nanomolar binding capacities of these two L-formed Immucillin analogues, plus they had the potential to be applied in different circumstances.
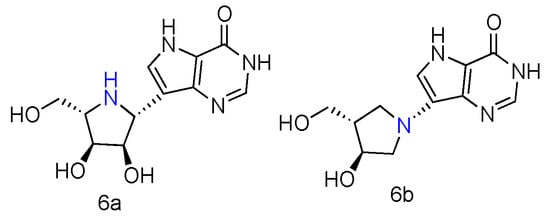
Figure 2. Structures of L-ImmH (6a) and L-DADMe-ImmH (6b).
4. L-d4T and L-ddC Derivatives as Anti-HIV Agents
There have been extensive studies in search of chemotherapeutics to effectively cure pathogenic Human Immunodeficiency Virus (HIV) [34]. The dominant experimental and clinical attempts are focused on the development of modified nucleoside analogues, which bind to HIV reverse transcriptase and interfere with the synthesis of DNA copying of the viral genome [35]. Successful examples include small molecule drugs of AZT [36], ddI [37], ddC [38], and d4T [39] that have been clinically used to treat AIDS patients. The lack of 2′- and 3′-hydroxyl groups in nucleoside sugar can cause the termination of HIV reverse transcription and inhibit the viral life cycle. In particular, d4T has the double bond in its pseudosugar ring to rigidify the ring to planar conformation. In order to engender more potent inhibitor against HIV with great activity and less cytotoxicity, a number of L-nucleoside have been reported, most of which bear the chemically derivatized pyrimidine and dideoxy L-ribose.
Some β-L-2′,3′-didehydro-2′,3′-dideoxythymidine (β-L-d4T) analogues (Figure 3, 7a) have been synthesized, all bearing a tether on the C-5 position of the uracil ring, and they were evaluated in vitro for anti-HIV-1 activity [40]. The results revealed that the L-d4T derivative, containing 12 methylene units at the 5-position, displayed some activity in the CEM-SS cells (IC50 2.3 µM), probably due to the more lipophilic nature of the nucleoside. Meanwhile, another innovative L-d4T derivative, L-MCd4T, containing the conformationally rigid methanocarba (MC) nucleoside, was synthesized, and its bicyclo[3.1.0]hexane moiety was restrained to North conformation and the 2′,3′-double bond further reduced the structural flexibility (Figure 3, 7b) [41]. L-MCd4T was found to be a potent anti-HIV-1 inhibitor (EC50 6.76 µg/mL) without significant cytotoxicity, which is comparable to clinical drug ddI [42].
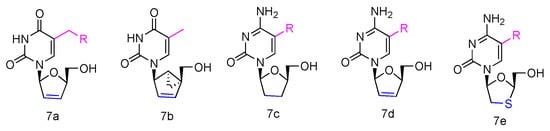
Figure 3. Structures of anti-HIV L-pyrimidine nucleoside analogues.
Additionally, some L-enantiomers of 2′,3′-dideoxycytidine analogues were reported to selectively inhibit HIV in various cell cultures. For example, L-2′,3′-dideoxycytidine, L-2′,3′-dideoxy-5-fluorocytidine (L-ddC and L-FddC, Figure 3, 7c), L-2′,3′-didehydro-2′,3′-dideoxycytidine, and L-2′,3′-didehydro-2′,3′-dideoxy-5-fluorocytidine (L-d4C and L-Fd4C, Figure 3, 7d) were found to have impressive inhibitory activity but significantly less toxicity when treated in different cells, including rat glioma, lung carcinoma, lymphoblastoid, and skin melanoma cells [43,44,45]. The L-enantiomers of 2′,3′-dideoxy-3′-thiacytidine and its 5-fluoro-derivative (L-3TC and L-FTC, Figure 3, 7e), when having a sulfur atom in place of the 3′-carbon, were discovered to be a potent inhibitor against HIV-1 in peripheral blood mononuclear cells and were also effective in thymidine kinase-deficient CEM cells. Meanwhile, nontoxicity was observed in human lymphocytes and other cell lines at up to 100 µM [46,47,48].
5. L-Azanucleoside as Anti-HBV Agents
Various azanucleoside analogues have been pioneered, in which the natural carbone atoms in nucleobases are substituted with bioisosteric nitrogens, and these innovative compounds exhibited promising antitumore activity [49,50]. Inspired by this observation, Sartorelli et al. synthesized various dioxolane azanucleosides with L-configuration (Figure 4, 8a–d) and bioevaluated them against HBV (Hepatitis B virus) [51]. The synthetic strategies involved the condensation of dioxolane derivative with silyl-protected azacytosine and azathymine. Interestingly, the in vitro HBV activity assay revealed that only (-)-(2S,4S)-1-[2-(hydroxy-methyl)-1,3-dioxolan-4-yl]-5-azacytosine (8a) possessed superior activity against HBV (EC50 = 0.6 µM), whereas its D-analogue was found inactive. No significant antiviral activity was observed in L-azathymidine analogues.
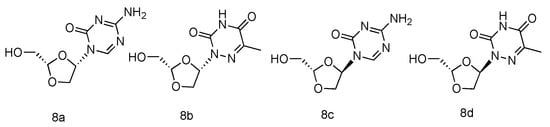
Figure 4. Structures of L-azanucleoside as an anti-HBV agent.
6. L-4′-Thionucleosides as Anti-Tumor Agents
The corresponding L-4′-thionucleosides (Figure 5, 9a–d) were synthesized by starting from 1,2,3,5-tetra-O-acetyl-4-thio-β-L-ribofuranose [52]. All tested compounds showed a moderate growth inhibitory activity against HTB14 human glioma cells. Notably, compounds 9b and 9c exhibited a significant growth stimulatory activity towards NB4 and T47D cells at concentrations 0.78–1.56 µM.
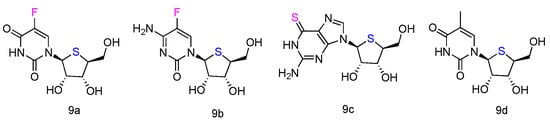
Figure 5. Structures of L-4′-thionucleosides.
7. L-5-Fluoronucleoside to Treat Leukemia
It has been reported that a type of cytidine analogue, Cytarabine or Ara-C, can be used as an effective chemotherapy against acute myeloid leukemia, acute lymphocytic leukemia, chronic myelogenous leukemia, and non-Hodgkin’s lymphoma [53]. Cytarabine has the arabinoside sugar to mimic the native deoxycytidine, and it is rapidly phosphorylated into cytosine arabinoside triphosphate (Ara-CTP) to interfere with the DNA synthesis in the S phase of the cancer cell cycle [54]. However, the drug resistance of Ara-C is quite common, because the effective Ara-CTP can be easily deaminated to an inactive uridine metabolite by cytidine deaminase (CDA) [55]. To address this issue, an L-nucleoside analogue of Ara-C, 5-fluorotroxacitabine (5FTRX) has been developed and activity tested in Acute Myeloid Leukemia (AML) cell lines [56].
5FTRX has an L-configuration, containing a fluorinated cytosine nucleobase and dioxolane ring as sugars (Figure 6, 10). Experimental results suggested that 5-FTRX could also be phosphorylated to its 5′-triphosphate nucleotide, leading to significant DNA chain termination during DNA replication and cancer cell death. In addition, compared to Ara-C, 5FTRX was observed to overcome the drug resistance induced by CDA overexpression to some extent. Meanwhile, no signs of significant toxicity were displayed in a mouse experiment.
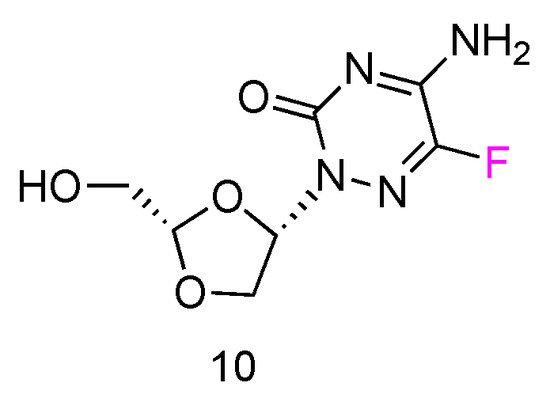
Figure 6. Structures of L-5-fluorotroxacitabine.
8. LdT as Anti-HBV Agent
The LdT drug, also known as Telbivudine and invented by Novartis Inc., is an FDA-approved (in 2006) anti-viral drug used in the treatment of hepatitis B infection (Figure 7, 11) [57]. Telbivudine has the L-converted structure of native thymidine, and it impairs the HBV DNA replication by leading to chain termination. Clinical trials have fully demonstrated its effect of viral suppression in patients and less viral resistance.
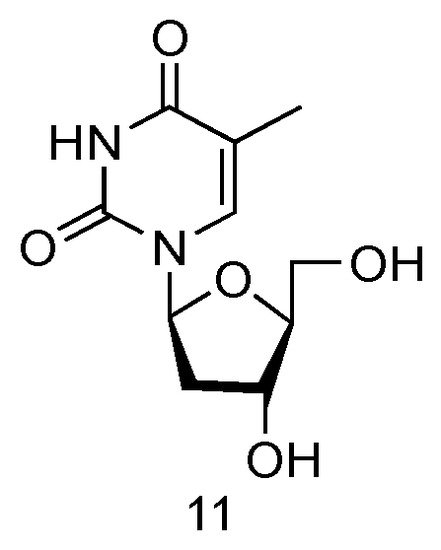
Figure 7. Structures of LdT.
9. 3TC and FTC to Treat HIV
Lamivudine and Emtricitabine (commonly called 3TC and FTC, Figure 8, 12a,b) are L-type cytidine analogues both containing oxathiolane rings as sugars. Emtricitabine has an additional Fluoro-modification at the 5-position of its cytosine nucleobase. Both compounds have been FDA approved for the treatment of human HIV and HBV infections, which can be administered individually or in combination with other inhibitors [58]. As the cytidine analogues, 3TC and FTC share the similar mechanism of action, by inhibiting the HIV reverse transcriptase and hepatitis B virus polymerase functions. The lack of 3′-OH groups prevents viral DNA elongation and terminates viral DNA growth. As the non-natural L-nucleoside, both drugs were identified as less toxic agents in mitochondrial DNA [59].
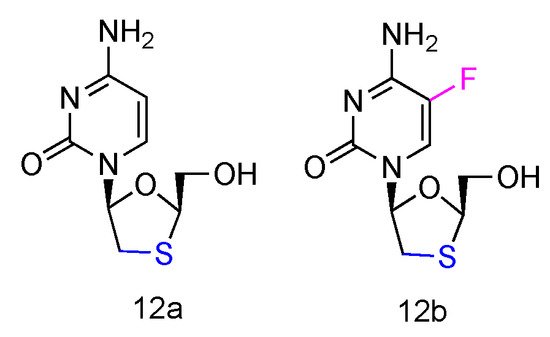
Figure 8. Structures of Lamivudine and Emtricitabine.
10. L-3′-Azido-2′,3′-dideoxypurine Nucleosides as Anti-HIV and Anti-HBV Agents
Inspired by the success of Lamivudine and Emtricitabine and the functional AZT-containing 3′-azido group, the Schinazi lab has prepared various L-3′-azido-2′,3′-dideoxypurine nucleosides (Figure 9, 13a,b), and evaluated their activity against HIV and HBV [60]. Eleven different L-nucleosides were obtained through microwave-assisted optimized transglycosylation reactions. These L-nucleoside analogues could be metabolized to corresponding nucleoside 5′-triphosphate compounds in primary human lymphocytes. Weak antiviral activities against HIV-1 and HBV were exhibited, even though no significant toxicity was observed.
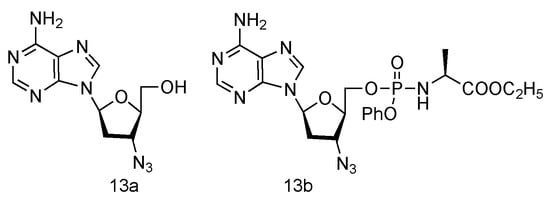
Figure 9. Structures of L-3′-azido adenosine and its phosphoramidate derivatives.
11. L-5′-Ethylenic and Acetylenic Modified Nucleosides
The unique 5′-cap structure in mRNA is essential for effective binding to ribosome [61]. Interference with the formation of cap structure could inhibit the replication process and provide the potential strategy for viral treatment. The 5′-cap formation relies on the catalysis by methyltransferases, which has become a popular target for anti-viral drug design [62]. It has been found that many adenosine analogues displayed the interesting antiviral activity by inhibiting S-Adenosyl-L-homocysteine (SAH) hydrolase, because SAH hydrolase is a key regulator of many S-adenosyl-L-methionine (SAM) dependent biological methylation processes [63]. Various 5′-ethylenic and acetylenic substituted L-adenosine derivatives were synthesized (Figure 10, 14a–e), and some of them showed modest inhibition of SAH hydrolase at 100 μM, when tested in the growth of HeLa cells or Bel-7420 cells [64].
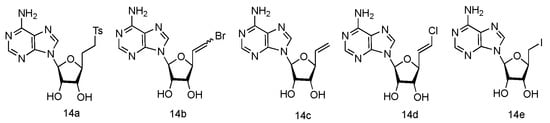
Figure 10. Structures of L-5′-ethylenic and acetylenic nucleosides.
12. L-3′-Cyano Modified Nucleosides
Following the similar principle of L-d4T and L-ddC to treat HIV by inhibiting the viral reverse transcription with reduced toxicity, the Chu lab has developed a series of L-nucleoside analogues containing a cyano group at the 3′-position (Figure 11, a–l). Some of the compounds also contained 2′,3′-unsaturated ribose [65]. The synthesized nucleosides were tested for anti-HIV activity in human PBM cells in vitro, and five of them (derivatives a, b, g, h and j) showed modest antiviral activity.
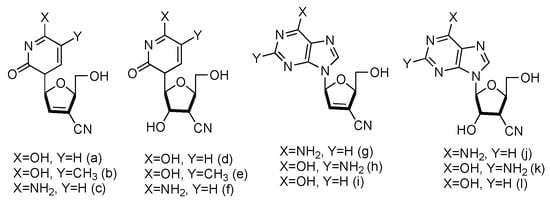
Figure 11. Structures of L-3′-cyano modified nucleosides.
13. L-Enantiomer of Ribavirin
Ribavirin is a nucleoside analogue used to treat respiratory syncytia viral infection [66] and HCV infection [67]. It is reported to have the effects of inducing type 1 cytokine bias and enhancing the T cell-demiated immunity in vivo [68]. Structurally, Ribavirin is similar to nucleoside, which has the nucleobase replaced by 1,2,4-triazole-3-carboxamide. Ramasamy et al. have synthesized a series of L-nucleoside analogues of Ribavirin and evaluated their activity of stimulating type 1 cytokine and enhancing T cell-demiated immunity [69]. One of the compounds prepared, which had 1,2,4-triazole-3-carboxamide (Figure 12, 17a) as its nucleobase, was found to be the most uniformly potent compound with interesting immunomodulatry potential.
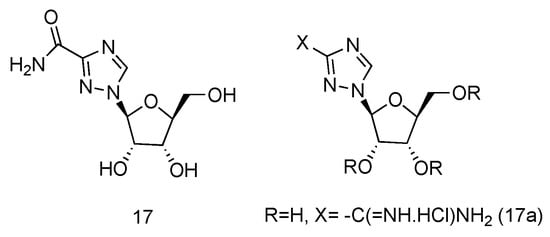
Figure 12. Structure of L-Ribavirin (17) and its derivative (17a).
14. L-Dideoxy Bicyclic Pyrimidine
Zika virus is a mosquito-born flavivirus that can cause the symptoms of fever, rash, joint pain, and red eyes [70]. Zika virus shares the same replication cycle as other flaviviruses, suggesting the potential of using nucleoside analogues as antiviral agents to terminate Zika virus DNA elongation. The Brancale lab has screened a targeted small molecule pool against Zika virus in vitro [71]. Several modified adenosine compounds have been identified to significantly inhibit the virus-induced cytopathic effect. Interestingly, one additional prodrug, 18a, which exhibited a L-nucleoside conformation, was also screened out (Figure 13). This analogue contained L-dideoxyribose sugar and a bicyclic pyrimidine as its nucleobase, and it had a potent synergistic effect of inhibiting the vaccinia and measles viruses when applied together with other adenosine phosphoramidate compounds.
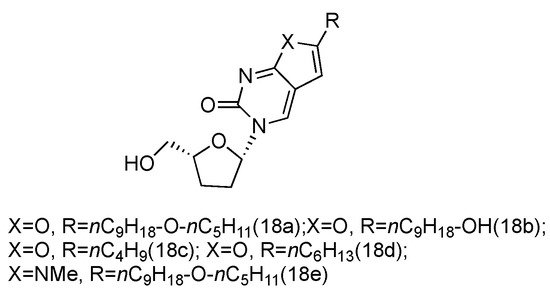
Figure 13. Structures of L-dideoxy bicyclic pyrimidine.
Above, researchers listed some of the L-nucleoside analogues with a solid demonstration of their therapeutic activities. Besides, there are many other nucleoside-like small molecules designed and evaluated as antiviral therapeutics, which had an L-configuration similar to its modified nucleoside, including cyclobutene L-nucleoside analogues [72], L-erythro-hexopyranosyl nucleosides [73,74], L-4′-C-ethynyl-2′-deoxypurine nucleosides [75], L-ribo-configured Locked Nucleic Acid [76,77,78,79], pyrrolo, pyrazolo, or imidazo-modified L-nucleoside [80], et al. There have also been many methods developed to efficiently synthesize carbocyclic L-nucleoside analogues [52,81,82,83], and they have been summarized elsewhere [84,85].
In the future development of L-nucleoside as therapeutic molecules, one direction is to design a broad-spectrum antiviral drug essential for rapid and efficient disease treatment. The recent viral outbreaks in the past decade have urged this need. Generally, the development of L-nucleosides with broad-spectrum antiviral activities is more challenging because of the different behaviors among viruses, especially after their infections to the host. To design a broad-spectrum antiviral nucleoside, a comprehensive investigation is needed to discover the biological features of multiple viruses and design chemically modified L-nucleosides to target these features. In addition, the combinatory treatment using different nucleoside drugs, or a nucleoside with another biological agent, will be necessary to decrease the drug resistance.
This entry is adapted from the peer-reviewed paper 10.3390/genes13010046
This entry is offline, you can click here to edit this entry!