Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Microbiology
|
Biochemistry & Molecular Biology
Pembroke JT and MP Ryan. Autothermal thermophilic aerobic digestion (ATAD) is a microbial fermentation process characterized as a tertiary treatment of waste material carried out in jacketed reactors. Heat is generated which selects a thermoduric microbial population. The process results in a stabilised, pasteurised sludge suitable for land application as a fertiliser. The microbial population biodegrades sludge contents, are unique in terms of diversity and have biotechnological potential as enzymes and proteins associated with the microbial population are thermostable.
- ATAD
- thermophilic aerobic sludge digestion
- Autothermal thermophilic Aerobic Digestion
- Stabilised Pasteurised Sludge
- Biofertiliser
1. Introduction
ATAD (autothermal aerobic thermophilic digestion) is a liquid composting, tertiary sludge treatment process which utilizes the metabolic heat generated by way of microbial digestion to stabilize the treated sludge and effectively pasteurize the digestate [1,2,3]. This produces class A biosolids suitable for land spread. The ATAD process has been utilized to treat a variety of waste streams including animal waste, food, brewery slaughterhouse wastes and domestic wastes. It has been demonstrated that ATAD can be used to process a variety of high strength waste streams at elevated temperatures with biological oxygen demand (BOD) reduction of 95% and chemical oxygen demand (COD) reductions up to 99% [4,5]. The process can operate as a one-stage system [6] or as a two-stage system [7] that incorporates a mesophilic stage and then follow on thermophilic stage. Generally, the sludge itself is thickened via polymer addition to 7–8% total solids followed by aeration in jacketed bioreactors, where the addition of air and mixing causes biodegradation to proceed with the energy converted to biomass and heat. Because of the jacketed nature of the bioreactors, the trapped heat results in a rise in temperature to above 60 °C, often reaching up to 75 °C [8]. The thermal processing results in a sanitized digestate [9] where pathogens, carried over from the initial secondary sludge, are inactivated. This results in a Class A Biosolids, which has the potential for land spread as a liquid fertilizer. During the process, stages deamination of proteinaceous material results in ammonia release [7]. This results in the case of domestic wastes, in a rise in pH to above 8. This increased pH has been proposed to allow better reactor performance, higher enzyme activity, facilitates oxidation, accelerates humification but limits microbial diversity by the unique combination of pH and temperature [10]. These conditions provide a unique environmental niche with a distinctive nutrient profile of often somewhat recalcitrant biological material that results from primary and secondary treatment waste processes the nature of which varies according to waste type.
2. Sampling of ATAD Diversity
The optimal determination of molecular diversity in an ATAD process, or indeed in any niche, relies heavily on the proper preparation and processing of microbial DNA from the sampling site. This target DNA is the key element, which is followed by subsequent analysis of the DNA by techniques such as 16S rDNA sequencing, denaturing gradient gel electrophoresis (DGGE) or sequencing for metagenome analysis. The validity of the subsequent phylogenies or dynamics of the populations recovered is highly dependent on the optimum recovery of the DNA. In attempting to carry out an analysis of the microbial diversity of a large scale ATAD reactor (110 m3) treating mixed domestic waste, a number of key limitations in the sampling and processing were identified [9]. These were not apparent in the analysis of more traditional sampling sites but were key to the proper analysis of the ATAD niche. Initial attempts to recovery the diversity of the microbial population indicated there were a number of factors that limited recovery. An extensive analysis of the conditions that limited diversity recovery was carried out in an attempt to optimize the identification of the key microbial players that participated in the ATAD process, particularly at the elevated temperatures [9,20,38]. During the various transition phases from mesophilic to thermophilic conditions, and also during the thermophilic stages, significant amounts of lysis were observed as the population transitioned to the increased temperatures or more alkaline conditions [33]. This resulted in the release of large amounts of nucleases with varied substrate specificities, many of which were thermostable and capable of interfering with the recovery of template DNA for subsequent phylogenetic analysis [20,38]. Additionally, this lead to the liberation of a diverse range of proteases, many of which were also thermostable were found to dramatically effect subsequent DNA amplification if not inhibited or removed during the initial DNA extraction protocols. Inhibition or removal of these activities was shown to be a key element of diversity recovery [20]. The presence of humic substances in the sludge was also shown to be inhibitory to a number of polymerases used to amplify templates for 16S rDNA analysis as were the choice of 16S primers, with variation shown in their ability to amplify the 16S rDNA target [38]. Analysis also determined that extraction methodology, choice of polymerase, and the use of additives during processing of DNA samples were all key to the optimal and reproducible recovery of ATAD microbial diversity [20,38]. Other strategies for optimizing recovery of environmental DNA were also examined, with many appearing in the literature these include serial dilution of the template [39], however this resulted in dilution also of rare templates which effects diversity recovery. Addition of a range of chemical additives [40] has also been proposed and trialed on ATAD DNA recovery [38], but these also depend on the nature of the inhibitory substances that might be present in ATAD samples. To verify the validity of ATAD DNA extracts to optimize diversity recovery three criteria used [20], the recovery of high molecular weight DNA, the absence of PCR inhibitors, and extracts that recover all possible genomes. A number of DNA extraction protocols were initially compared, spike and recovery of known templates was carried out, the quality of the DNA analyzed and the reactivity of the extracted template DNA to a variety of DNA polymerases and 16S rDNA primers analyzed to optimize the recovery and subsequent molecular analysis of ATAD DNA [18]. Examination indicated that a number of factors associated with ATAD sludge caused interference with optimal recovery of DNA. These included the presence of humic substances in ATAD sludge, where even small quantities have been shown to inhibit DNA polymerase activities [41]. The presence of nucleases, which are liberated during lysis as the temperatures increase, needed addition of EDTA at 5 mM (some five times recommended in buffers) and formamide, and heat treatment of the extracted DNA was necessary to limit template and even primer degradation [14,20,38]. Dilution of template DNA was found to relieve PCR inhibition but this had a major effect on diversity recovery. Neither did the efficiency of PCR recovery correlate with the length of the PCR template. A number of 16S rDNA primers were trialed, but no correlation was detected with recovery and amplicon length. It was observed that use of V3-V3 and V1-V19 16S rDNA primer sets were more resistant to inhibition with ATAD extracts, and these were routinely utilized [20]. Given that many other ATAD substances, in addition to humic substances, present in ATAD sludge such as carbohydrates and fiber could affect amplification a number of adjuvants were also utilized in attempts to optimize amplification. It was observed that the addition of bovine serum albumen (BSA), (which may bind polyphenols and humic substances and act as alternative substrates for residual extracted ATAD proteases) and formamide (weakens hydrogen bonds between nucleotides reducing complex formation and enhances specificity, possesses some DNase inhibitory activity) resulted in better DNA recovery for further analysis. Spiked additions of diluted, known templates were added periodically as a control to the ATAD extracts to determine the ability of the optimized techniques to recovery ‘rare’ (diluted) spiked DNA [20]. This aided in the validation of the various adjuvant additions for optimizing the template purification strategy. In summary, key elements identified [20,38] were that specific modifications to extraction and amplification reactions were necessary to optimize recovery of templates for molecular analysis of ATAD sludge microbial communities. The Power Soil DNA isolation kit (Qiagen, Manchester, UK) was found to give the best extraction coupled to the modifications specific to ATAD as outlined above [20]. A number of other key elements were also identified, such as processing the samples immediately following sampling as storage or transport was found to limit template DNA recovery, while maintaining DNA in 1% formamide was essential for longer-term storage. Thus, when attempting to determine the true community diversity of an unusual niche, such as ATAD at elevated temperatures, optimizing the preparation protocols is an essential element in recovery of this diversity.
3. Phylogenetic Analysis of the ATAD Community at the Thermophilic Stage
The ATAD process is used globally and it is well recognized that the microbial community present contributes to the degradation of total solids in ATAD sludge, to sludge ecology and to heat production [5,48]. There have been relatively few studies on the phylogeny of organisms present in ATAD reactors at the thermophilic stage. Such molecular studies have been restricted to ATAD systems treating swine waste [21] and pharmaceutical wastes [17] and interestingly some anaerobes, mainly Clostridia were identified in addition to aerobes. In terms of ATAD, treating domestic waste materials the first report focused on a waste mixture from a combined domestic activated sludge mixed with meat processing waste [19]. Here, the V3 region of bacterial and archaeal 16S rDNA was amplified from extracted DNA. No archael species were detected as confirmed later [14].
Using ATAD optimized extraction and purification strategies for domestic waste sludge [20,38], a comprehensive phylogenetic study was carried out on an ATAD treating mixed domestic waste sludge [2]. To limit PCR bias, amplification was carried our using universal primers 27F and 1492R to amplify almost the complete 16S rDNA gene using hot start, low cycle number with five replicas and utilizing touchdown PCR [49]. These were purified and used to generate a comprehensive clone library in the pGEM-T Easy vector (Promega, Southampton, UK), Clones demonstrating unique patterns via Amplified rDNA Restriction Analysis (ARDRA) were chosen, examined by DGGE and those deemed unique were reamplified using V3-V5 region primers. These unique sequences representing optimal ATAD diversity were sequenced and utilized for bioinformatics analysis [2]. All rDNA sequences obtained were deposited in the GenBank database under accession numbers GU376459-GU376467, GU348989, GU325824-GU325838, GU320653-GU320666, and GU437223-GU437232. Some 400 clones were examined with some 27 operational taxonomic units (OTUs) identified by ARDRA analysis. Fifty-four near full-length 16S rDNA amplicons, two each for each of the 27 OTUs were utilized. Most of the resulting clones demonstrated >92% sequence identity to deposited sequences in the Ribosomal Database. Representatives of High GC Actinobacteria, low GC Gram-positive Firmicutes, as well as α and β- Proteobacteria were observed. 22 of the phylotypes detected grouped within the Firmicutes with some fifteen of the Phylotypes representing Clostridia species indicating that although termed ‘aerobic’ significant anaerobic metabolism may be a feature of ATAD, perhaps due to poor mixing, poor oxygen solubility at elevated temperatures and the rate of oxygen utilization at elevated temperatures (Figure 1). Many clones demonstrated similarity to compost related uncultured organisms, which had previously been reported [17,19] from other ATAD studies. The predominance of the Firmicutes was identified in all studies and is also a feature of later phylogenetic studies [3]. Similarity to known organisms was demonstrated [14] with Bacillus thermocloaceae 98% (clone ER66), Anoxybacillus 99% (clone ER17), to Ureibacillus 99% (clone F27) and the alkaline tolerant Bacillus infernus 99% (clone CK81). ATAD Clones ER33, CK36, CK85, CK40, CK17, CK3, CK34, CK29, and CK8 showed between 96–99% similarity to cultured and uncultured Clostridia species. Clone ER22 and ER34 showed similarity to Symbiobacterium, clone ER9 to Moorella, while clone CK10 showed similarity to a Dehalobacter species. Clone ER19 showed 97% similarity to Rhodobacter, while clone ER58 showed similarity to Sphingomonadaceae both belonging to the α-Proteobacteriae. Clone CK11 was related to Aquamonas belonging to the β-Proteobacteria. Two phylotypes, CK46 and CK31, showed similarity to Actinobacteria (Microbacterium and Tetrashpaera respectively). No clones related to Thermus were recovered in this study as reported elsewhere [19,50] but this may reflect differences in the waste as a substrate source, the reactor design, or the operating parameters (Figure 2).
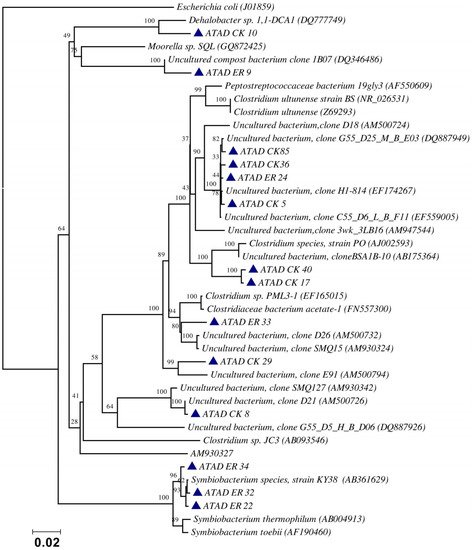
Figure 1. Evolutionary relationship of the ATAD clones and their closest neighbors shown as a phylogenetic tree among the Firmicutes, class Clostridia and relatives of the domain Bacteria derived by neighbor-joining analysis of 16S rDNA sequences. The bootstrap consensus tree inferred from 1000 replicates is taken to represent the evolutionary history of the taxa analyzed [51]. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown above the branches. The evolutionary distances were computed using the maximum composite likelihood method and are in the units of the number of base substitutions per site. There were a total of 1238 positions in the final dataset. Phylogenetic analyses were conducted in MEGA 4.0 [52]. Reproduced with permission from Water Research, Elsevier.
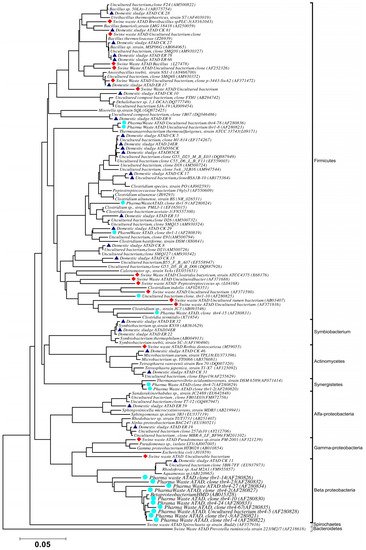
Figure 2. Phylogenetic positioning and evolutionary relationship of 16S rDNA clones and isolated strains obtained from ATAD reactors and their nearest neighbors Accession numbers of the sequences retrieved from GenBank are given together with their names. ATAD isolates from domestic waste [2] are illustrated with closed triangle (blue), isolates and clones from ATAD treated swine manure [21] (closed rectangular orange), isolates and clones form ATAD treated pharmaceutical waste (closed circle aquamarine) [17]. Bar scale represents 0.05 nucleotide substitutions per position. The evolutionary relationship was inferred using the neighbor-joining method, with evolutionary distances computed using the maximum composite likelihood method [53] and are in the units of number of base substitutions per site. Phylogenetic analyses were conducted in MEGA 4.0 [52]. Reproduced with permission from Water Research, Elsevier.
Using a Japanese ATAD as a model, Tashiro et al. [3] carried out principal-coordinate analysis of 16S rDNA amplicons from some 454,122 sequencing reads from 20 different sampling regimes during the ATAD process. They reported a predominance of Bacteroides and Firmicutes with Proteobacteria showing a dramatic increase as the process initiated. As the process increased in temperature Bacteroides numbers increased while Proteobacteria decreased. As the temperature increased towards the thermophilic stage Acinobacteria increased and the Firmicutes became the dominant phylum [3]. This unique limited species similarity was also illustrated in an examination of an ATAD generating liquid fertilizer in Japan where during the thermophilic phase a stable bacterial community was observed with major OTUs showing limited similarities to Heliorestis baculata, Caldicellulosiruptor bescii, and Ornatilinea apprima [23]. As with previous studies, the assignment to nearest genera showed only 84% similarity indicative that many of the bacterial species have so far not been isolated or identified. This picture is similar to previous studies [2,12,17,19,50] where OTU assignment to closest relative ranged from between 80–95% for many of the 16S rDNA species detected. This is suggestive that there are many new species to be yet identified and cultured, particularly at the thermophilic stages. Because many of these species may be thermophilic or thermoduric, live at high population densities and in at alkaline pH there are biotechnological opportunities to discover potential antibiotics (which may control access to substrate at dense populations) and thermostable enzymes with unique specificities. These may be able to utilize more recalcitrant substrates present during the late thermophilic stages with potentially a wider spectra of pH tolerances.
Although it may be impossible to remove all bias, strategies to optimize molecular approaches for determining ATAD community structure at elevated temperatures have aided in recovering this diversity [2,3,17,19,21]. The data suggests a restricted phyla distribution but a richness in members of the Firmicutes, suggesting an important role for them in ATAD digestion. However data suggests that ATAD community structure may reflect unique players depending on the chemical composition of the waste being treated and the process operating conditions with variation observed depending on the waste whether swine waste [21], pharmaceutical waste [17], meat waste [19], or mixed domestic waste [2]. While no Bacillaceae were detected in pharmaceutical ATAD waste, these were found to be particularly rich in all other studies reported. The presence of anaerobes also appears to be important with anaerobes related to Clostridia being detected in all published studies. However, their detection does not necessarily mean that they are active metabolically, a question that needs to be addressed in future studies.
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms7080215
This entry is offline, you can click here to edit this entry!