The etiology of chicken muscular dystrophy is the synthesis of aberrant WWP1 protein made by a missense mutation of WWP1 gene. The β-dystroglycan that confers stability to sarcolemma was identified by Cho et al. as a substrate of WWP protein, which induces the next molecular collapse. The dystrophin-glycoprotein complex (DGC) is a core protein of costamere that is an essential part of force transduction and protects the muscle fibers from contraction-induced damage. Caveolin-3 (Cav-3) and dystrophin bind competitively to the same site of β-dystroglycan, and excessive Cav-3 on sarcolemma will block the interaction of dystrophin with β-dystroglycan, which is another reason for the disruption of the DGC. It is known that fast-twitch glycolytic fibers are more sensitive and vulnerable to contraction-induced small tears than slow-twitch oxidative fibers under a variety of diseased conditions. Accordingly, the fast glycolytic αW fibers must be easy with rapid damage of sarcolemma corruption seen in chicken muscular dystrophy, but the slow oxidative fibers are able to escape from these damages.
- caveolin-3
- chicken muscular dystrophy
- WWP1
- beta-dystroglycan
- stretching
- fiber types
- combined transplantation
- myotube formation
- mitochondria
1. Introduction
In 1954, an animal model with inherited muscular dystrophy was found in a commercial flock of New Hampshire chickens. The poultry farmers brought affected birds to the department of poultry science (later changed to avian science) at the University of California at Davis. Drs. Asmundson and Julian, in the school of veterinary medicine, examined them clinically and pathologically. The early results were reported by Asmundson and Julian [1].
Fertilized eggs from this source were used to establish a line homozygous for the abnormality. The most consistent clinical symptom was caused by the disfunction of the fast-twitch muscles with a predominance of fast-twitch αW and fast-twitch αR fibers [2], which correspond to the Type IIB and Type IIA fibers in mammalian muscles, respectively [3]. Pectoralis muscle (M. pectoralis superficialis) from breast part, PLD muscle (M. posterior latissimus dorsi) from the dorsal part and biceps muscle (M. biceps brachii) and patagialis muscle (M. tensor patagiii longus) from the fore arm and the patagium of the upper arm, respectively, are examples of chicken fast-twitch muscles which revealed severe pathological changes after hatching (Figure 1b,c). Among fast-twitch muscles, pectoralis muscle was the earliest and most severely affected soon after 2–3 weeks ex ovo. Chickens flap their wings using these muscles for flying up and right themselves instantly from the spine position when placed on their back [4]. Birds that could rise five times are assigned a score of 6 and considered normal. As the disease progresses, they fail to rise due to the inability to move breast and wing muscles. This movement performance is designated as flip test or exhaustion test to evaluate the disease progression and pharmaceutical effect (Figure 1a).

Figure 1. Chickens with muscular dystrophy (line 413) cannot right themselves from the spine position when they placed on their back while normal birds stand up instantly from this position (a). The pectoralis muscles from normal (b) and dystrophic (c) chickens at seven months are stained with Sirius Red. Normal pectoral muscle fibers (line 412) have polygonal contour and yellow cytoplasm outlined basal lamina by bright red line. They are wrapped by reddish connective tissue. Dystrophic pectoralis muscles are earliest and most severely affected, which are characterized by a marked variation in size with a proliferation of intracellular nuclei, necrotic phagocytosis (arrow), multivesicular fibers (arrow head) and fibrosis with lipid droplets. Note that dystrophic fibers lead to develop thicker endomysium layer compared to age matched wild-type ones. Bars in (b) and (c) indicate 80 and 100 μm, respectively.
On the other hand, the mixed-fiber type muscles include, in addition to the fast-twitch fibers, the slow-twitch βR fibers or few slow-tonic α’ and β’ fibers. They localize mostly in the leg, neck and dorsal positions, and they are capable of retaining relatively normal state for months after hatching. The twitch fibers of chicken have a single motor end plate, whereas slow-twitch βR and slow-tonic fibers are multiply innervated [5][6]. The complexus muscle (M. complexus) from the neck is a mixed type, which contains, among the majority of fast-twitch fibers, a few slow-twitch βR or slow-tonic α’ or β’ fibers. The former corresponds to Type I fibers and the latter belongs to Type IIIA or IIIB fibers in mammals, respectively (Table 1) [3][7].
Table 1. Classification of different fiber types in chicken muscle. The twitch muscle fibers with high myofibrillar adenosine triphosphatase (ATPase) after alkaline preincubation and low activity after acid preincubation are called α fibers. They are divided further into two subtypes, αW fibers with low oxidative activity and αR fibers with high oxidative activity (succinic dehydrogenase, SDH) or nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR). The fibers with the reverse ATPase and high oxidative activity are called βR fibers. The tonic muscle is also heterogeneous in fiber types with respect to ATPase activity, which do not reverse in myofibrillar ATPase pattern when exposed to acidic or alkaline preincubation. The β’ fibers with more intense reaction tend to locate centrally among groups of lighter-staining α’ fibers in a similar way in twitch muscles, which consist of βR fibers located centrally within groups of α fibers. The classification in mammals was made by Brook and Keizer [8]. The multiple innervation of βR fibers in twitch muscles was reported for the first time by Ashmore et al. (1978) [5][6].
| Muscle Fiber Types | Twitch Fibers | Tonic Fibers | |||
|---|---|---|---|---|---|
| Ashmore and Doerr (1971) [2] | αW | αR | βR | α’ | β’ |
| Brooke and Kaiser (1970) [8] | ||B | ||A | | | |||A | |||B |
| Histochemical criteria | |||||
| ATPase (pH 10) | ● | ● | ○ | ● | ● |
| ATPase (pH 4.1) | ○ | ○ | ● | ◎ | ● |
| SDH or NADH-TR | ○ | ◎ | ● | ◎ | ● |
| Phosphorylase | ● | ◎ | ○ | ◎ | ◎ |
| Innervation pattern | Focal | Focal | Multiple | Multiple | Multiple |
The ALD muscle (M. anterior latissimus dorsi) from the dorsal anterior position contains almost entirely slow-tonic fibers and responds to denervation with marked hypertrophy, but its tension developed in response to the application of K+-rich solutions is reduced by about 50% [9]. The sartorius, adductor and iliofibularis muscles from the leg are mixed-type composed of five types of fibers, αW, αR, βR and two types of slow-tonic fibers. In contract, posterior iliotibialis muscle contains predominantly fast-twitch fibers [10].
In the cross section of normal pectoralis muscles, many polygonal muscle fibers are bundled together and wrapped in a thin layer of connective tissue covering. Most of the muscle nuclei are located at the periphery of each fiber (Figure 1b). A marked pathological change in dystrophic pectoralis muscles are: a hypertrophy of muscle mass with fiber size variation, a proliferation of myofiber nuclei and satellite cells, cytoplasmic vacuolization, necrotic destruction of muscle fibers and the presence of ring fibers and fibrosis with fatty infiltration where muscle fibers are replaced by connective tissue [11][12][13][14][15] (Figure 1c).
The inheritance pattern was initially thought as an autosomal recessive mode designated as abnormal muscle (am/am) for homozygotes [1][11]. However, because dystrophic phenotypes are often seen in heterozygous muscles, they came to be identified as a co-dominant inheritance designated a gene symbol AM/AM [16][17]. Chicken muscular dystrophy is transmitted by a single gene, but the phenotype is modified by other background genes [16][17][18][19]. One of several lines of dystrophic New Hampshire chickens, line 413, was introduced with normal line 412 from the University of California at Davis to Japan in 1976 [17].
A traditional approach to the gene function sets about a phenotype analysis approach to a gene that encodes the phenotype. Before 1980, until advances in DNA technology based on the positional cloning and reverse genetics, very few human genes had been identified as disease loci. Despite the accumulation of experimental data obtained by these studies, the etiology of Duchenne muscular dystrophy has been unidentified for many years.
The reverse genetic approach clarifies abnormal cellular functions and various disease phenotypes from gene mutation. For example, the genetic analysis and positional cloning revealed a dystrophin gene mutation responsible for the Duchenne muscular dystrophy in 1987 [20]. The positional cloning for the genetic screenings opened new avenues to identify the gene mutation responsible for the chicken muscular dystrophy. Matsumoto et al. (2008) identified a WWP1 gene mutation that led to an arg441-to-glu (R441Q) substitution in chickens with inherited muscular dystrophy [21]. The WW domain containing E3 ubiquitin protein ligase 1 (WWP1) is one of the ubiquitin ligases which play an important role in ubiquitin-proteasome pathway.
2. Caveolin-3: Another Causative Process of Muscular Dystrophy
Caveolae are flask-shaped vesicular invaginations of plasma membrane which are known to regulate endocytosis, exocytosis, cholesterol homeostasis, signal transduction and mechanoprotection [22]. Caveolin-3 (Cav-3) is the structural protein component of caveolae in skeletal and cardiac muscle cells [23][24] and promotes proper clustering of AChRs but diffuses the distribution of AChRs in myotubes deficient Cav-3 [24][25]. Although Cav-3 is predominantly associated with sarcolemma of mature muscles, it distributes with the transverse tubule (T-tubule) system in differentiating myotubes [26][27]. The T-tubules of Cav-3 null mice are dilated and run in irregular directions, suggesting that Cav-3 is involved in the organization of T-tubules but not essential for their formation [28]. Cav-3 deficiency induces a muscular dystrophic phenotype (Rippling disease) [29], while its overexpression does harm to muscle tissue [30][31][32]. Cav-3 overexpressing transgenic mice revealed severely affected symptoms with an increase in the number of sarcolemmal caveolae, which is one of the Duchenne muscular dystrophy traits in humans, and the downregulation of dystrophin and β-dystroglycan protein expression [31].
Moreover, these mice show elevated blood serum creatine kinase (CK) levels and are consistent with the marked elevation of this enzyme in chicken muscular dystrophy [33]. In addition, serum pyruvate kinase (PK) activities in dystrophic chickens were approximately 30-fold higher than those in normal chickens, while a seven-fold elevation was detected in serum CK activity at 37 days of age (Table 2) [17]. It is thought that the rise of serum CK and PK causes lysis or necrosis of muscle fibers, with subsequent release of these enzymes into the blood [16]. The caveolae function in buffering and resealing mechanical stresses at the plasma membrane, including sarcolemma and T-tubules system [34]. The freeze-fracture morphology of sarcolemma in soleus muscle in Cav-3 −/−, Cav-3 +/− and Cav-3 +/+ wild-type mice revealed that caveolae were abundant in wild-type mice, less frequent in heterozygous Cav-3 +/− mice and scarce in Cav-3 −/− mice [29]. The pectoralis muscles were investigated in normal and dystrophic chickens at embryo, early post-hatching and adult stages [35]. The average densities of caveolae are higher (30 /μm2) in adult dystrophic fibers than those (17 /μm2) in age-matched normal fibers. The distribution pattern of caveolae in dystrophic fibers is random arrangement compared to rectangular one in normal fibers. These abnormalities were already obvious at seven days after hatching before the appearance of clinical symptoms. In contrast, the sarcolemma of slow tonic ALD fibers have randomly dispersed caveolae whose appearance and distribution are unaffected throughout the life by this myopathy [36]. Matsumoto et al. (2010) reported the expression of Cav-3 and other caveolae-related proteins in adult normal and dystrophic chickens (Figure 2a). Western blotting and semi-quantitative RT-PCR analysis revealed that Cav-3 is higher only in affected fast-twitch muscles of dystrophic chickens and the amount of caveolin-3 protein is regulated in posttranslational modification, since no significant increase is observed at the mRNA level (Figure 2b).
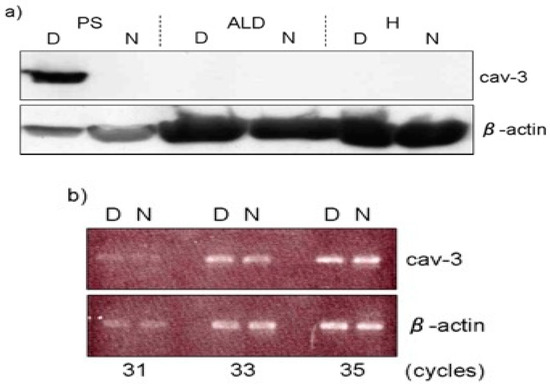
Figure 2. Expression of caveolin-3 (Cav-3) at protein and mRNA level. (a) Expression of Cav-3 in M. pectoralis superficialis (PS), M. anterior latissimus dorsi (ALD) and heart (H) was analyzed by Western blotting. Note that PS expressed higher amount of Cav-3 protein (7.12 ± 3.31-fold) in dystrophic chickens (D), while the expression in ALD and H was undetectable as in normal chickens (N). (b) The semi-quantitative RT-PCR analysis indicated that its mRNA expression was at the similar level between dystrophic (D) and normal (N) pectoralis muscle. Adopted from Matsumoto et al. 2010 [32].
Table 2. Blood serum pyruvate kinase (PK) and creatine phosphokinase (CPK) activities in normal, dystrophic and heterozygous carrier chicks a.
| Enzyme | Age (Days) | Normal b (mU/mL) | Dystrophy b (mU/mL) | Heterozygote b (mU/mL) |
|---|---|---|---|---|
| PK | 37 | 401 ± 111(8) | 12,430 ± 6,269(4) c | 630 ± 209(5) |
| 70–86 | 405 ± 73(4) | 10,773 ± 6,800(7) c | 1,213 ± 552(10) d | |
| 475 | 229 ± 33(7) | 8,940 ± 4,032(5) c | 516 ± 138(7) d | |
| CPK | 37 | 141 ± 55(8) | 1,071 ± 812(4) c | 180 ± 31(5) |
| 70–86 | 164 ± 41(4) | 1,146 ± 599(7) c | 183 ± (10) | |
| 475 | 30 ± 9(7) | 986 ± 586(5) c | 46 ± 10(7) e |
Cav-3 and dystrophin bind to the same site of β-dystroglycan and interact competitively with β-dystroglycan [37]. It was reported that WWP1, which is a primary causative protein of chicken muscular dystrophy, also requires its site in β-dystroglycan. Cav-3 and dystrophin compete against WWP1 as well to negatively affect WWP1-mediated β-dystroglycan degradation [38]. Accordingly, these results explain why the overexpression of Cav-3 induces the destabilization and degradation of the DGC complex, leading to major defects in membrane integrity and intracellular myofibril alignments.
As mentioned above, caveolae play a role in buffering mechanical stress at the plasma membrane, since muscle fibers are repeatedly experiencing mechanical stress at the plasma membrane, which must be able to rapidly repair wounds. The failure to repair causes the degenerative changes in Z-band and myofibrils in embryonic pectoralis muscle fibers [39]. McLean et al. (1986) also found extensive changes in patterning of sarcolemmal caveolae of chicken dystrophic PLD muscle, but the patterning of normal fibers is arranged in striking bands over the myofibrillar I-bands [36]. These morphological changes in caveolae structure and distribution are already detected in dystrophic pectoralis fibers as early as seven days after hatching [35]. Together, results indicate that the overexpression of Cav-3 protein is involved in another causative process of chicken muscular dystrophy.
The primary cause of chicken muscular dystrophy is due to an aberrant WWP1 protein which targets β-dystroglycan as substrate. The β-dystroglycan is a core molecule of the DGC and guarantees stability to sarcolemma [40]. Loss of β-dystroglycan in dystrophic fast-twitch fibers are vulnerable to contraction-induced wounding and are likely to undergo repeated cycles of injury and repair. Caveolae provide reserve force of expandable membrane when tension is added. Stretching of muscle caused a loss of caveolae, apparently by their flattening [41], and a similar effect, “unfolding” of caveolae, was proposed in endothelial cells upon changes in capillary volume [42]. Caveolae were shown to flatten in response to changes in membrane tension, both upon cell swelling or with stretch. This process was energy-independent and caused release of cavins from the caveolae, raising the possibility that cavins may act as cytosolic signals for changes in membrane tension [43].
Feit et al. (1985 and 1989) measured both tension and stiffness as a function of muscle length under relaxing conditions on isolated small bundles of chemically skinned pectoralis myofibers from normal and dystrophic chickens aged between 45 and 55 days. They indicated that dystrophic pectoralis muscles show increased proportions of high-molecular-weight collagen, suggestive of increased cross-linking and are stiffer than normal muscles, and develop more tension for the same amount of stretch [44][45].
Fujii, Murota and Tanzer (1983) found an increase in amount of total collagen with an increased proportion of Type III collagen in muscle as early as 13 days following progressive deterioration to 19 days of embryos. This suggests the production of more immature collagen fibers compared to normal ones. The stiffness is mediated by such altered form of collagen which is collagenase-resistant by virtue of excessive crosslinking [46]. Moreover, an ultrastructural study of the tendon in embryonic gastrocnemius muscles showed significant alterations in developing myotendinous junction from dystrophic chickens as early as 13 days of embryos [47].
Then, it is of interest to elucidate what happen if dystrophic muscles with considerable amount of Type III collagen fibers in extracellular space and deteriorated myofibrils in immature myotubes, are added the tension to some extent for various periods? The stretch-induced growth in chicken wing patagialis muscles were conducted by Ashmore’s group during three years after 1980. The patagialis muscles, one of the fast twitch muscles, were extended at six weeks of age for 1–5 weeks. The passive stretch is a powerful inducer of muscle growth and the sift of fiber types from αW to αR fibers as the percent of αR fibers had increased from 11% in the control to 43% in the stretched muscles [48][49]. It was also confirmed in rat and rabbit that a chronic increase in tension as a result of stretch applied by the lengthening procedure is a potent stimulus for fast-to-slow myosin transformation and for muscle hypertrophy [50][51].
The patagialis muscles of normal and dystrophic chickens at seven days ex ovo were stretched for six weeks [52]. The stretched dystrophic muscles increased in weight, muscle mass cross-sectional area and fiber cross-sectional area, revealing a protective effect of stretch against the progressive pathology of muscular dystrophy. Stretched dystrophic muscles contained a higher rate of slow-twitch αR fibers than normal stretched muscles. The dystrophic fibers showed a more striking hypertrophy and have a larger number of mitochondria with intense SDH activity than those in age-matched normal muscles, so that they were a more uniform oxidative fiber profile compared with normal fibers stretched.
As mentioned earlier, the combined transplantation of both normal and dystrophic muscle fragments produces a single hybrid myofiber in which normal and dystrophic nuclei coexist. The regenerating fibers indicate regional differences in oxidative enzyme and growth rate along their length [53]. These results were supported by the histochemical reaction of SDH activity in longitudinal sections of combined transplantation. Although it is not clear whether the fiber hypertrophy and mitochondrial SDH activity in cytoplasm are programmed genetically, or occur as a secondary compensatory response, it appears that either of these changes is characteristic of stretched and transplanted muscles. It has been suggested that mitochondria from dystrophic pectoralis muscles have not only significantly higher concentration but also higher basal activity stabilizing to greater degree than normal pectoralis muscle [54].
From these points of view, it was revealed that β-dystroglycan ubiquitinated by excessive afferent WWP1 causes an accumulation of caveolin, stiffness and tension due to extracellular space with immature collagens and distorted myofibrils in dystrophic muscles beginning from in ovo stages. In addition, Lee et al. (2013) reported in a C2C12 cells in vitro study that WWP1 protein interacts with AMP-activated protein kinase (AMPK) and downregulates its expression through ubiquitin ligase activity in skeletal muscle [55]. The AMPK is a sensor of cellular energy change, maintains the energy balance by decreasing the ATP-consuming processes and associates with increased mitochondrial enzyme content and mitochondrial biogenesis in rat skeletal muscle [56][57]. The immunoactivity of WWP1 antibody to sarcolemma in dystrophic pectoralis fibers is weaker than in control pectoralis fibers, whereas dystrophic fibers contain mitochondrial signals distributed much more densely compared to those of normal pectoralis muscles [58]. This result might be associated with increased mitochondria and their higher enzyme activity in dystrophic fibers compared to those in normal fibers. It is necessary to prove whether abnormally ubiquitinated AMPK and related proteins in dystrophic chickens would increase necessary protein levels via a fiber type shift, resulting in more slow, oxidative fibers that are much more resistant to contraction-induced damage.
In 2005, the hypoglycosylation and laminin-binding of defective α-dystroglycan were implicated to trigger the onset of chicken muscular dystrophy [59]. However, there were no proteins associated to glycosylation in the AM region on chicken chromosome 2q and aberrant glycosylation does not appear to be caused by its direct interaction with mutated WWP1 protein. The short sugar chain of α-dystroglycan in dystrophic muscles might be an early phase of the secondary result of the pathological changes following deregulation of β-dystroglycan on sarcolemma.
This entry is adapted from the peer-reviewed paper 10.3390/biom10091206
References
- Asmundson, V.S.; Julian, L.M. Inherited muscle abnormality in the domestic fowl. J. Hered. 1956, 47, 248–252, doi:10.1093/oxfordjournals.jhered.a106655.
- Ashmore, C.R.; Doerr, L. Postnatal development of fiber types in normal and dystrophic skeletal muscle of the chick. Expl Neurol. 1971.30, 431-446. Doi :10.1016/0014-4886(78)90140-1
- Barnard, E.A.; Lyles, J.M.; Pizzey, J.A. Fibre types in chicken skeletal muscles and their changes in muscular dystrophy. J. Physiol. 1982, 331, 333–354, doi:10.1113/jphysiol.1982.sp014375.
- Julian, L.M. Animal model: Hereditary muscular dystrophy of chickens. Am. J. Pathol. 1973, 70, 273–276. PMID: 4566993 PMCID: PMC1903970.
- Ashmore, C.R..; Kikuchi, T.; Doerr, L. Some observations on the innervation patterns of different fiber types of chick muscle. Exp. Neurol. 1978, 58, 272–284, doi:10.1016/0014-4886(78)90140-1.
- Ashmore, C.R..; Vigneron, P.; Marger, L.; Doerr, L. Simultaneous cytochemical demonstration of muscle fiber types and acetylcholinesterase in muscle fibers of dystrophic chickens. Exp. Neurol. 1973, 60, 68–82, doi:10.1016/0014-4886(78)90169-3.
- Ashmore, C.R.; Addis, P.B.; Doerr, L.; Stokes, H. Development of muscle fibers in the complexus muscle of normal and dystrophic chicks. J. Histochem. Cytochem. 1973, 21, 266–278, doi:10.1177/21.3.266.
- Brooke, M.H.; Kaiser, K.K. Muscle Fiber Types: How Many and What Kind? Arch. Neurol. 1970, 23, 369–379, doi:10.1001/archneur.1970.00480280083010.
- Kikuchi, T.; Schmidt, H. Changes in resting and contractile properties of chicken muscles following denervation. Biomed. Res. 1983, 4, 303–314, doi:10.2220/biomedres.4.303.
- Rafuse, V.F.; Milner, L.D.; Landmesser, L.T. Selective Innervation of Fast and Slow Muscle Regions during Early Chick Neuromuscular Development. J. Neurosci. 1996, 16, 6864–6877, doi:10.1523/JNEUROSCI.16-21-06864.1996.
- Asmundson, V.S.; Kratzer, F.H.; Julian, L.M. Inherited myopathy in the chicken. Ann. N. Y. Acad. Sci.1966, 138, 49–58, doi:10.1111/j.1749-6632.1966.tb41153.x.
- Holliday, T.A.; Julian, L.M.; Asmundson, V.S. Muscle growth in selected lines of muscular dystrophic chickens. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1968, 160, 207–216, doi:10.1002/ar.1091600207.
- Randall, W.R..; Wilson, B.W. Properties of muscles from chickens with inherited muscular dystrophy. J. Neurol. Sci. 1980, 46, 145–155, doi:10.1016/0022-510x(80)90073-8.
- Nonaka, I.; Sugita, H. Intracytoplasmic vacuoles in αW fibers of dystrophic chicken muscle—Probable early pathologic event initiates massive fiber necrosis. Acta Neuropathol. 1981, 55, 173–181, doi:10.1007/bf00691315.
- Ohwada, S.; Kikuchi, T. A microphotometric study on the proliferation pattern of muscle cell nuclei in chickens with hereditary muscle dystrophy. Tohoku J. Agric. Res. 1985, 36, 63–68. http://hdl.handle.net/10097/29861
- Wilson, B.W.; Randall, W.R.; Patterson, G.T.; Entrikin, R.K. Major physiologic and histochemical characteristics of inherited dystrophy of the chicken. Ann. N. Y. Acad. Sci. 1979, 317, 224–246, doi:10.1111/j.1749-6632.1979.tb56531.x.
- Kikuchi, T.; Ishiura, S.; Nonaka, I.; Ebashi, S. Genetic heterozygous carriers in hereditary muscular dystrophy of chickens. Tohoku J. Agric. Res. 1981, 32, 14–26. http://hdl.handle.net/10097/29802.
- Kikuchi, T.; Moriya, H.; Matsuzaki, T.; Katoh, M.; Takeda, S. The Development of Laboratory Animal Science for the Study of Human Muscular and Nervous Diseases in Japan. Congenit. Anom. 1987, 27, 447–462, doi:10.1111/j.1741-4520.1987.tb00726.x.
- Kondo, K.; Kikuchi, T.; Mizutani, M. Breeding of the chick as an animal model for muscular dystrophy. In Proceedings of the International Symposium on Muscular Dystrophy, Tokyo, Japan, 25–27 November 1980; pp. 19–24.
- Koenig, M.; Hoffman, E.; Bertelson, C.; Monaco, A.P.; Feener, C.; Kunkel, L. Complete cloning of the duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517, doi:10.1016/0092-8674(87)90504-6.
- Matsumoto, H.; Maruse, H.; Inaba, Y.; Yoshizawa, K.; Sasazaki, S.; Fujiwara, A.; Nishibori, M.; Nakamura, A.; Takeda, S.; Ichihara, N.; et al. The ubiquitin ligase gene (WWP1) is responsible for the chicken muscular dystrophy. FEBS Lett. 2008, 582, 2212–2218, doi:10.1016/j.febslet.2008.05.013.
- Thomas, C.M.; Smart, E.J. Caveolae structure and function. J. Cell. Mol. Med. 2008, 12, 796–809.
- Song, K.S.; Scherer, P.E.; Tang, Z.; Okamoto, T.; Li, S.; Chafel, M.; Chu, C.; Kohtz, D.S.; Lisanti, M.P. Expression of Caveolin-3 in Skeletal, Cardiac, and Smooth Muscle Cells. Caveolin-3 Is a Component of the Sarcolemma and Cofractionates with Dystrophin and dystrophin -associated glycoproteins. J. Biol. Chem. 1996, 271, 15160–15165.
- Hagiwara, Y.; Nishina, Y.; Yorifuji, H.; Kikuchi, T. Immunolocalization of caveolin-1 and caveolin-3 in monkey skeletal, cardiac and uterine smooth muscles. Cell Struct. Funct. 2002, 27, 375–382.
- Hezel, M.; De Groat, W.C.; Galbiati, F. Caveolin-3 Promotes Nicotinic Acetylcholine Receptor Clustering and Regulates Neuromuscular Junction Activity. Mol. Biol. Cell 2010, 21, 302–310.
- Parton, R.G.; Way, M.; Zorzi, N.; Stang, E. Caveolin-3 Associates with Developing T-tubules during Muscle Differentiation. J. Cell Biol. 1997, 136, 137–154.
- Ralston, E.; Ploug, T. Caveolin-3 Is Associated with the T-Tubules of Mature Skeletal Muscle Fibers. Exp. Cell Res. 1999, 246, 510–515.
- Galbiati, F.; Engelman, J.A.; Volonte, D.; Zhang, X.L.; Minetti, C.; Li, M.; Hou, H.; Kneitz, B.; Edelmann, W.; Lisanti, M.P. Caveolin-3 Null Mice Show a Loss of Caveolae, Changes in the Microdomain Distribution of the Dystrophin-Glycoprotein Complex, and T-tubule Abnormalities. J. Biol. Chem. 2001, 276, 21425–21433.
- Hagiwara, Y.; Sasaoka, T.; Araishi, K.; Imamura, M.; Yorifuji, H.; Nonaka, I.; Ozawa, E.; Kikuchi, T. Caveolin-3 deficiency causes muscle degeneration in mice. Hum. Mol. Genet. 2000, 9, 3047–3054.
- Galbiati, F.; Volonté, D.; Minetti, C.; Bregman, D.B.; Lisanti, M.P. Limb-girdle Muscular Dystrophy (LGMD-1C) Mutants of Caveolin-3 Undergo Ubiquitination and Proteasomal Degradation. J. Biol. Chem. 2000, 275, 37702–37711.
- Galbiati, F.; Volonté, D.; Chu, J.B.; Li, M.; Fine, S.W.; Fu, M.; Bermudez, J.; Pedemonte, M.; Weidenheim, K.M.; Pestell, R.G.; et al. Transgenic overexpression of caveolin-3 in skeletal muscle fibers induces a Duchenne-like muscular dystrophy phenotype. Proc. Natl. Acad. Sci. USA 2000, 97, 9689–9694.
- Matsumoto, H.; Sasazaki, S.; Fujiwara, A.; Ichihara, N.; Kikuchi, T.; Mannen, H. Accumulation of caveolin-3 protein is limited in damaged muscle in chicken muscular dystrophy. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2010, 157, 68–72.
- Weinstock, I.; Behrendt, J.; Jones, K. Pyruvate kinase and creatine phosphokinase during development of the chicken with muscular dystrophy. Life Sci. 1977, 21, 1199–1205.
- Echarri, A.; Del Pozo, M.A. Caveolae-mechanosensitive membrane invaginations linked to actin filaments. J. Cell Sci. 2015, 128, 2747–2758.
- Costello, B.R.; Shafiq, S.A. Freeze-fracture study of muscle plasmalemma in normal and dystrophic chickens. Muscle Nerve 1979, 2, 191–201.
- McLean, B.; Mazen-Lynch, L.; Shotton, D.; McLean, G.A. Quantitative freeze?fracture studies of membrane changes in chicken muscular dystrophy. Muscle Nerve 1986, 9, 501–514.
- Sotgia, F.; Lee, J.K.; Das, K.; Bedford, M.; Petrucci, C.; Macioce, P.; Sargiacomo, M.; Bricarelli, F.D.; Minetti, C.; Sudol, M.; et al. Caveolin-3 directry interact with the C-terminal tail of β-dystroglycan. J. Biol. Chem. 2000, 275, 38048–38058.
- Cho, E.-B.; Yoo, W.; Yoon, S.K.; Yoon, J.-B. β-dystroglycan is regulated by a balance between WWP1-mediated degradation and protection from WWP1 by dystrophin and utrophin. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 2199–2213.
- Allen, E.R.; Murphy, B.J. Early detection of inherited muscular dystrophy in chickens. Cell Tissue Res. 1979, 197, 165–167.
- Cohn, R.D.; Campbell, K.P. Molecular basis of muscular dystrophies. Muscle Nerve 2000, 23, 1456–1471.
- Dulhunty, A.F.; Franzini-Armstrong, C. The relative contributions of the folds and caveolae to the surface membrane of frog skeletal muscle fibres at different sarcomere lengths. J. Physiol. 1975, 250, 513–539.
- Lee, J.; Schmid-Schönbein, G.W. Biomechanics of skeletal muscle capillaries: Hemodynamic resistance, endothelial distensibility, and pseudopod formation. Ann. Biomed. Eng. 1995, 23, 226–246.
- Sinha, B.; Köster, D.V.; Ruez, R.; Gonnord, P.; Bastiani, M.; Abankwa, D.; Stan, R.V.; Butler-Browne, G.; Védie, B.; Johannes, L.; et al. Cells Respond to Mechanical Stress by Rapid Disassembly of Caveolae. Cell 2011, 144, 402–413.
- Feit, H.; Kawai, M.; Schulman, M.I. Stiffness and contractile properties of avian normal and dystrophic muscle bundles as measured by sinusoidal length perturbations. Muscle Nerve 1985, 8, 503–510.
- Feit, H.; Kawai, M.; Mostafapour, A.S. The role of collagen crosslinking in the increased stiffness of avian dystrophic muscle. Muscle Nerve 1989, 12, 486–492.
- Fujii, K.; Murota, K.; Tanzer, M.L. Abnormal collagen synthesis in skeletal muscle of dystrophic chicken. Biochem. Biophys. Res. Commun. 1983, 111, 933–938.
- Sweeny, P.R. Ultrastructure of the developing myotendinous junction of genetic dystrophic chickens. Muscle Nerve 1983, 6, 207–217.
- Holly, R.G.; Barnett, J.G.; Ashmore, C.R.; Taylor, R.G.; Molé, P.A. Stretch-induced growth in chicken wing muscles: A new model of stretch hypertrophy. Am. J. Physiol. Physiol. 1980, 238, C62–C71.
- Barnett, J.G.; Holly, R.G.; Ashmore, C.R. Stretch-induced growth in chicken wing muscles: Biochemical and morphological characterization. Am. J. Physiol. Physiol. 1980, 239, C39–C46.
- Loughna, P.T.; Izumo, S.; Goldspink, G.; Nadal-Ginard, B. Disuse and passive stretch cause rapid alterations in expression of developmental and adult contractile protein genes in skeletal muscle. Development 1990, 109, 217–223.
- Pattullo, M.C.; Cotter, M.A.; Cameron, N.E.; Barry, J.A. Effects of lengthened immobilization on functional and histochemical properties of rabbit tibialis anterior muscle. Exp. Physiol. 1992, 77, 433–442.
- Ashmore, C.R. Stretch-induced growth in chicken wing muscles: Effects on hereditary muscular dystrophy. Am. J. Physiol. Physiol. 1982, 242, C178–C183.
- Kikuchi, T.; Doerr, L.; Ashmore, C.R. A possible mechanism of phenotypic expression of normal and dystrophic genomes on succinic dehydrogenase activity and fiber size within a single myofiber of muscle transplants. J. Neurol. Sci. 1980, 45, 273–286.
- Ashmore, C.R.; Doerr, L. Oxidative metabolism in skeletal muscle of normal and dystrophic chicks. Biochem. Med. 1970, 4, 246–259.
- Lee, J.O.; Lee, S.K.; Kim, N.; Kim, J.H.; You, G.Y.; Moon, J.W.; Jie, S.; Kim, S.J.; Lee, Y.W.; Kang, H.J.; et al. E3 ubiquitin ligase, WWP1, Interacts with AMPKα2 and down-regulates its expression in skeletal muscle C2C12 cells. J. Biol. Chem. 2013, 288, 4673–4680.
- Winder, W.W.; Holmes, B.F.; Rubink, D.S.; Jensen, E.B.; Chen, M.; Holloszy, J.O. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J. Appl. Physiol. 2000, 88, 2219–2226.
- Bergeron, R.; Ren, J.M.; Cadman, K.S.; Moore, I.K.; Perret, P.; Pypaert, M.; Young, L.H.; Semenkovich, C.F.; Shulman, G.I. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Metab. 2001, 281, E1340–E1346.
- Imamura, M.; Nakamura, A.; Mannen, H.; Takeda, S. Characterization of WWP1 protein expression in skeletal muscle of muscular dystrophy chickens. J. Biochem. 2016, 159, 171–179.
- Saito, F.; Blank, M.; Schröder, J.; Manya, H.; Shimizu, T.; Campbell, K.P.; Endo, T.; Mizutani, M.; Kröger, S.; Matsumura, K. Aberrant glycosylation of α-dystroglycan causes defective binding of laminin in the muscle of chicken muscular dystrophy. FEBS Lett. 2005, 579, 2359–2363.